リキッドバイオプシーワークフロー向けメチル化検出の強化
リキッドバイオプシー技術は、がんの早期検出やモニタリングのあり方を急速に変革しています。低侵襲なアプローチを提供するリキッドバイオプシーでは、cfDNA(セルフリー DNA)などの循環バイオマーカーを解析でき、がんに関連する遺伝的・エピジェネティック的変化に関する貴重な情報を得られます。そうしたバイオマーカーの中でも DNA メチル化の変化は、高い特異性と感度をもってさまざまながんを特定・追跡できる有望な手段として注目されています。
しかし、cfDNA は存在量が限られ、特有の断片化パターンを示すため、利用には独自の課題が伴います。cfDNA 断片の大半は長さが約 167 bp で、ヌクレオソームに由来することを反映しています。この断片化により、以下の問題が生じます。
- 断片の開始位置・終了位置の多様性の低さにより、実際はユニークな分子であっても重複リードとして扱われる傾向が増える
- ライブラリの複雑性が低下し、アッセイの感度が損なわれる可能性がある
これらの固有の特性によって従来のメチル化検出ワークフローでは cfDNA 解析が十分に行えず、これらの制約に対応するための高度なツールや技術が求められています。
Twist メチル化 UMI アダプターは、分子バーコード(UMI)を用いた正確な重複除去を可能にし、低多様性のサンプルにおける誤ったDuplicate 判定を減らすことで、cfDNA のメチル化ワークフローを向上させます。そのアダプターは EM-Seq プロトコルとの親和性を考慮した設計となっており、断片長を維持しつつ使用可能なデータを最大化して、ターゲットのカバレッジと再現性を向上させます。そのため、高い信頼性が要求されるメチル化研究に欠かせないツールとなります。

リキッドバイオプシーワークフロー向けメチル化検出の強化
リキッドバイオプシー技術は、がんの早期検出やモニタリングのあり方を急速に変革しています。低侵襲なアプローチを提供するリキッドバイオプシーでは、cfDNA(セルフリー DNA)などの循環バイオマーカーを解析でき、がんに関連する遺伝的・エピジェネティック的変化に関する貴重な情報を得られます。そうしたバイオマーカーの中でも DNA メチル化の変化は、高い特異性と感度をもってさまざまながんを特定・追跡できる有望な手段として注目されています。
しかし、cfDNA は存在量が限られ、特有の断片化パターンを示すため、利用には独自の課題が伴います。cfDNA 断片の大半は長さが約 167 bp で、ヌクレオソームに由来することを反映しています。この断片化により、以下の問題が生じます。
- 断片の開始位置・終了位置の多様性の低さにより、実際はユニークな分子であっても重複リードとして扱われる傾向が増える
- ライブラリの複雑性が低下し、アッセイの感度が損なわれる可能性がある
これらの固有の特性によって従来のメチル化検出ワークフローでは cfDNA 解析が十分に行えず、これらの制約に対応するための高度なツールや技術が求められています。
Twist メチル化 UMI アダプターは、分子バーコード(UMI)を用いた正確な重複除去を可能にし、低多様性のサンプルにおける誤ったDuplicate 判定を減らすことで、cfDNA のメチル化ワークフローを向上させます。そのアダプターは EM-Seq プロトコルとの親和性を考慮した設計となっており、断片長を維持しつつ使用可能なデータを最大化して、ターゲットのカバレッジと再現性を向上させます。そのため、高い信頼性が要求されるメチル化研究に欠かせないツールとなります。
UMI の導入でさらにDuplicate解析を改善
UMI を用いた duplicate判定により、ライブラリのduplicate率が 15% 以上減り、標準的な位置に基づくduplicate判定と比べてライブラリの複雑性が 25% ~ 35%、平均ターゲットカバレッジが 6% ~ 8% 向上します。(図1)
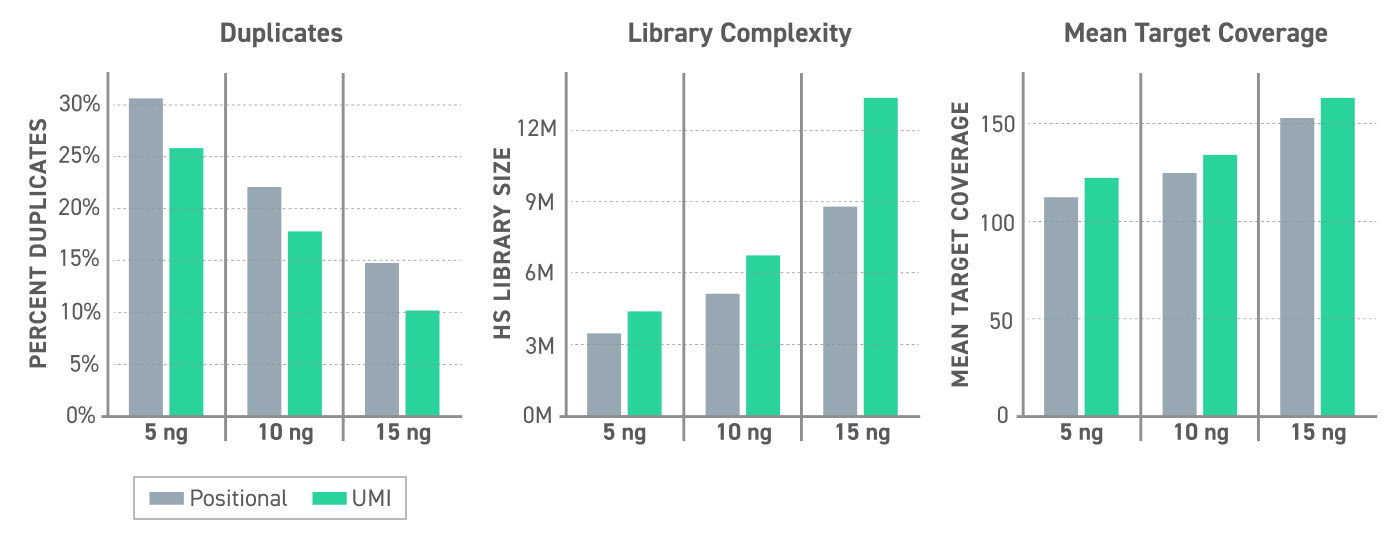
図 1:さまざまな cfDNA インプット量における UMI の性能
NEBNext EM-Seq ライブラリ調製に続いて、cfDNA を 5、10、または 15 ng 投入し、Twist メチル化 UMI アダプターを用いて EM-seq 変換ライブラリを作製しました。ライブラリはターゲットメチル化シーケンシングプロトコルを用いて Twist Alliance Pan-cancer Methylation Panel - 1.5MB でキャプチャし、Illumina Nextseq(550X ~ 1000X の生データカバレッジ)でシーケンシングしました。Duplicate リードの判定は、標準的な位置ベースの方法である GATK MarkDuplicates ツール、または UMI を用いた GATK UmiAwareMarkDuplicatesWithMateCigar ツールで行いました。
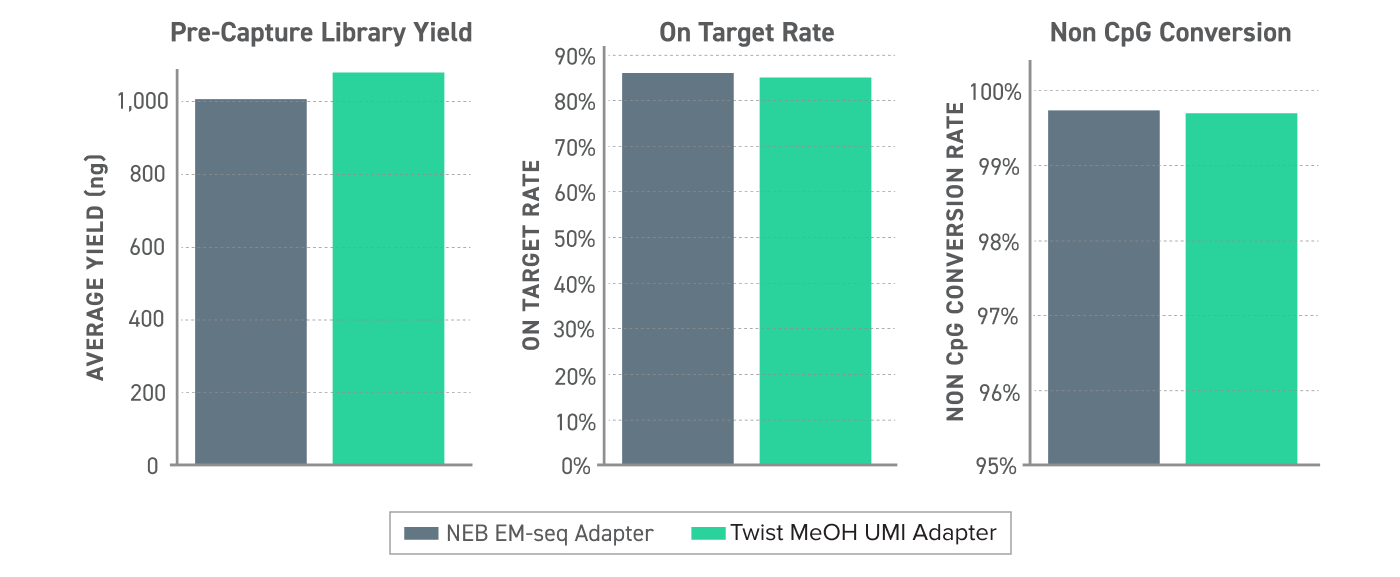
図 2:Twist メチル化検出ワークフローにおけるメチル化 UMI の性能
ヒト cfDNA を 10 ng 使用して、NEB EMseq アダプターまたは 新しい Twist メチル化 UMI アダプターを用い、EM-seq 変換ライブラリを作製しました。ライブラリは、ターゲットメチル化シーケンシングプロトコルを用いて Twist Alliance Pan-cancer Methylation Panel - 1.5MB でキャプチャし、Illumina Nextseq(550X ~ 1000X の生データカバレッジ)でシーケンシングしました。
バイオインフォマティクスワークフローの例
UMI 情報はバイオインフォマティクスのパイプラインに組み込み、下流解析に先立ってDuplicate リードを除去するために活用できます。

図 3:重複マーキングのために UMI を処理する解析ワークフローの概要
生リードはアダプター配列のマーキングと UMI 情報の抽出のために処理されます。続いて BWA-meth を用いて、リファレンスゲノムに対してリードのアライメントを行い、UMI 情報を使って重複マーキングを行います。重複マーキング後は、GATK を用いてPicard 指標を収集し、追加の解析が可能です。
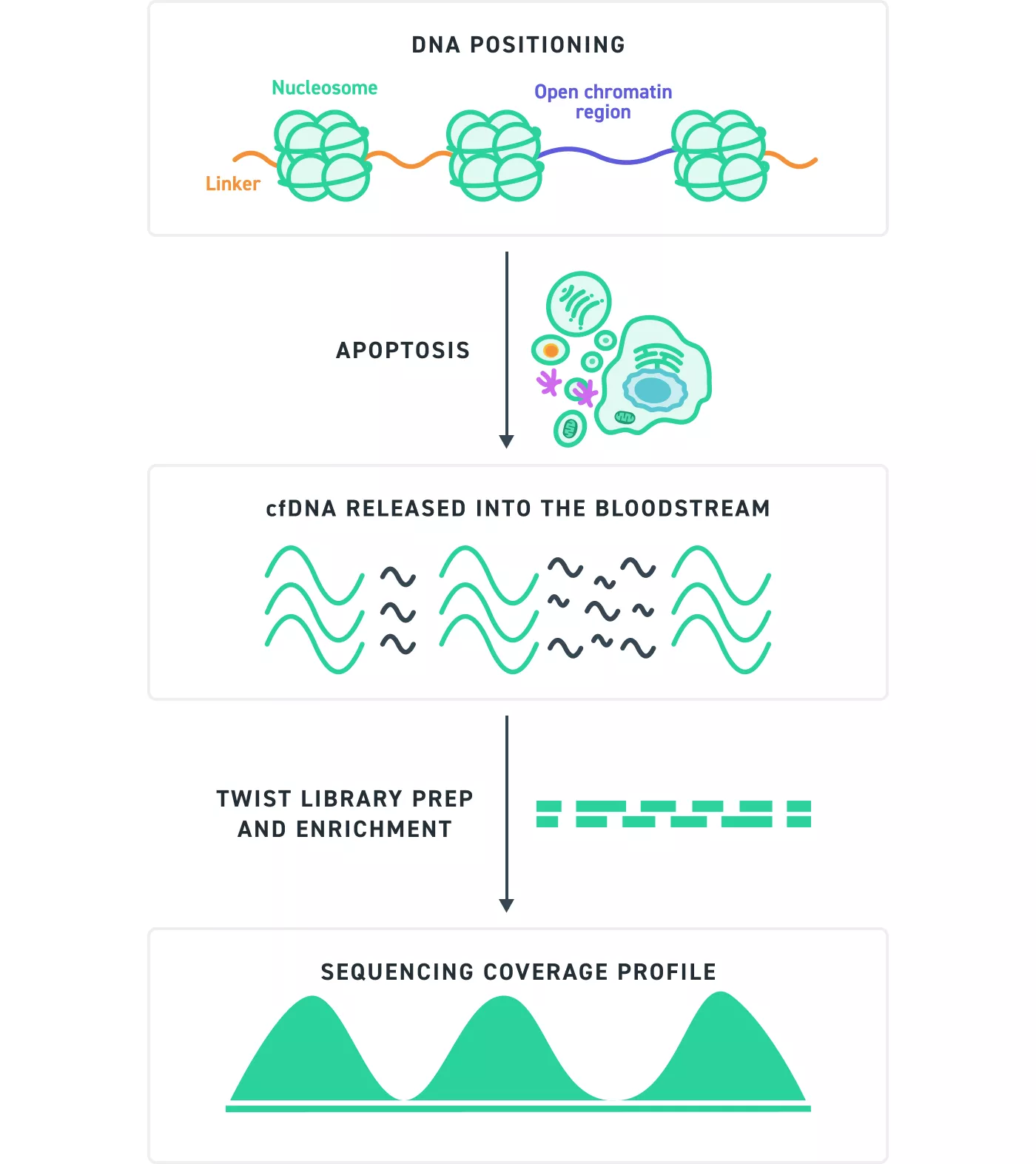
図 4:アポトーシスによる cfDNA 断片化からシーケンシングカバレッジまでの模式図:
アポトーシスの過程で、セルフリー DNA(cfDNA)は血流中に放出されます。エンドヌクレアーゼが DNA をランダムに断片化する一方で、DNA はヌクレオソームに巻き付くため位置的バイアスが生じ、断片の開始・終了位置における多様性が制限されます。その結果、本来は異なる分子であっても、シーケンシングデータ上ではduplicate として扱われる場合があります。ライブラリ調製時に分子バーコード(UMI)を付加することで、各分子を個別にラベル化でき、真の重複なのか別の分子なのかを正確に区別できます。
UMI の導入でさらにDuplicate解析を改善
UMI を用いた duplicate判定により、ライブラリのduplicate率が 15% 以上減り、標準的な位置に基づくduplicate判定と比べてライブラリの複雑性が 25% ~ 35%、平均ターゲットカバレッジが 6% ~ 8% 向上します。(図1)
図 1:さまざまな cfDNA インプット量における UMI の性能
NEBNext EM-Seq ライブラリ調製に続いて、cfDNA を 5、10、または 15 ng 投入し、Twist メチル化 UMI アダプターを用いて EM-seq 変換ライブラリを作製しました。ライブラリはターゲットメチル化シーケンシングプロトコルを用いて Twist Alliance Pan-cancer Methylation Panel - 1.5MB でキャプチャし、Illumina Nextseq(550X ~ 1000X の生データカバレッジ)でシーケンシングしました。Duplicate リードの判定は、標準的な位置ベースの方法である GATK MarkDuplicates ツール、または UMI を用いた GATK UmiAwareMarkDuplicatesWithMateCigar ツールで行いました。
図 2:Twist メチル化検出ワークフローにおけるメチル化 UMI の性能
ヒト cfDNA を 10 ng 使用して、NEB EMseq アダプターまたは 新しい Twist メチル化 UMI アダプターを用い、EM-seq 変換ライブラリを作製しました。ライブラリは、ターゲットメチル化シーケンシングプロトコルを用いて Twist Alliance Pan-cancer Methylation Panel - 1.5MB でキャプチャし、Illumina Nextseq(550X ~ 1000X の生データカバレッジ)でシーケンシングしました。
バイオインフォマティクスワークフローの例
UMI 情報はバイオインフォマティクスのパイプラインに組み込み、下流解析に先立ってDuplicate リードを除去するために活用できます。
図 3:重複マーキングのために UMI を処理する解析ワークフローの概要
生リードはアダプター配列のマーキングと UMI 情報の抽出のために処理されます。続いて BWA-meth を用いて、リファレンスゲノムに対してリードのアライメントを行い、UMI 情報を使って重複マーキングを行います。重複マーキング後は、GATK を用いてPicard 指標を収集し、追加の解析が可能です。
図 4:アポトーシスによる cfDNA 断片化からシーケンシングカバレッジまでの模式図:
アポトーシスの過程で、セルフリー DNA(cfDNA)は血流中に放出されます。エンドヌクレアーゼが DNA をランダムに断片化する一方で、DNA はヌクレオソームに巻き付くため位置的バイアスが生じ、断片の開始・終了位置における多様性が制限されます。その結果、本来は異なる分子であっても、シーケンシングデータ上ではduplicate として扱われる場合があります。ライブラリ調製時に分子バーコード(UMI)を付加することで、各分子を個別にラベル化でき、真の重複なのか別の分子なのかを正確に区別できます。
110830
製品:Twist Methylated UMI Adapters - TruSeq Compatible, 96 Sample110830
製品:Twist Methylated UMI Adapters - TruSeq Compatible, 96 Sample 製品シート
製品シート

