Detección mejorada de metilación para flujos de trabajo de biopsia líquida
Las tecnologías de biopsia líquida están transformando rápidamente el panorama de la detección y el seguimiento temprano del cáncer. Al ofrecer un enfoque mínimamente invasivo, las biopsias líquidas permiten el análisis de biomarcadores circulantes como el cfDNA (ADN libre de células), que contienen información valiosa sobre las alteraciones genéticas y epigenéticas asociadas al cáncer. Entre estos biomarcadores, los cambios en la metilación del ADN se destacan como una vía prometedora para identificar y rastrear diversos tipos de cáncer con alta especificidad y sensibilidad.
A pesar de su potencial, trabajar con ADNlc presenta desafíos únicos debido a su abundancia limitada y sus patrones de fragmentación distintos. La mayoría de los fragmentos de ADNlc tienen una longitud de aproximadamente 167 pb, lo que refleja su origen unido al nucleosoma. Esta fragmentación da como resultado:
- Baja diversidad de inicio y parada, lo que da como resultado moléculas más únicas llamadas lecturas de secuenciación duplicadas
- Reducción de la complejidad de la biblioteca, lo que puede dificultar la sensibilidad del ensayo.
Estas propiedades inherentes hacen que los flujos de trabajo tradicionales de detección de metilación sean menos efectivos para el análisis de ADNlc, lo que requiere herramientas y técnicas avanzadas diseñadas para abordar estas limitaciones.
Los adaptadores UMI metilados de Twist mejoran los flujos de trabajo de metilación de ADNlc al permitir una deduplicación precisa a través de identificadores moleculares únicos (UMI), lo que reduce las llamadas de duplicación falsa en muestras de baja diversidad. Su diseño garantiza la compatibilidad con los protocolos EM-Seq, conservando la longitud de los fragmentos, maximizando los datos utilizables y dando como resultado una mejor cobertura y reproducibilidad del objetivo, lo que los convierte en una herramienta fundamental para estudios de metilación de alta confianza.

Detección mejorada de metilación para flujos de trabajo de biopsia líquida
Las tecnologías de biopsia líquida están transformando rápidamente el panorama de la detección y el seguimiento temprano del cáncer. Al ofrecer un enfoque mínimamente invasivo, las biopsias líquidas permiten el análisis de biomarcadores circulantes como el cfDNA (ADN libre de células), que contienen información valiosa sobre las alteraciones genéticas y epigenéticas asociadas al cáncer. Entre estos biomarcadores, los cambios en la metilación del ADN se destacan como una vía prometedora para identificar y rastrear diversos tipos de cáncer con alta especificidad y sensibilidad.
A pesar de su potencial, trabajar con ADNlc presenta desafíos únicos debido a su abundancia limitada y sus patrones de fragmentación distintos. La mayoría de los fragmentos de ADNlc tienen una longitud de aproximadamente 167 pb, lo que refleja su origen unido al nucleosoma. Esta fragmentación da como resultado:
- Baja diversidad de inicio y parada, lo que da como resultado moléculas más únicas llamadas lecturas de secuenciación duplicadas
- Reducción de la complejidad de la biblioteca, lo que puede dificultar la sensibilidad del ensayo.
Estas propiedades inherentes hacen que los flujos de trabajo tradicionales de detección de metilación sean menos efectivos para el análisis de ADNlc, lo que requiere herramientas y técnicas avanzadas diseñadas para abordar estas limitaciones.
Los adaptadores UMI metilados de Twist mejoran los flujos de trabajo de metilación de ADNlc al permitir una deduplicación precisa a través de identificadores moleculares únicos (UMI), lo que reduce las llamadas de duplicación falsa en muestras de baja diversidad. Su diseño garantiza la compatibilidad con los protocolos EM-Seq, conservando la longitud de los fragmentos, maximizando los datos utilizables y dando como resultado una mejor cobertura y reproducibilidad del objetivo, lo que los convierte en una herramienta fundamental para estudios de metilación de alta confianza.
Incorporación de UMI en el análisis para mejorar la resolución de duplicados
La llamada duplicada informada por UMI reduce los duplicados de la biblioteca en más de un 15 %, lo que genera un aumento en la complejidad de la biblioteca (25-35 %) y la cobertura media del objetivo (6-8 %) en comparación con la llamada duplicada basada en posición estándar. (Figura 1)
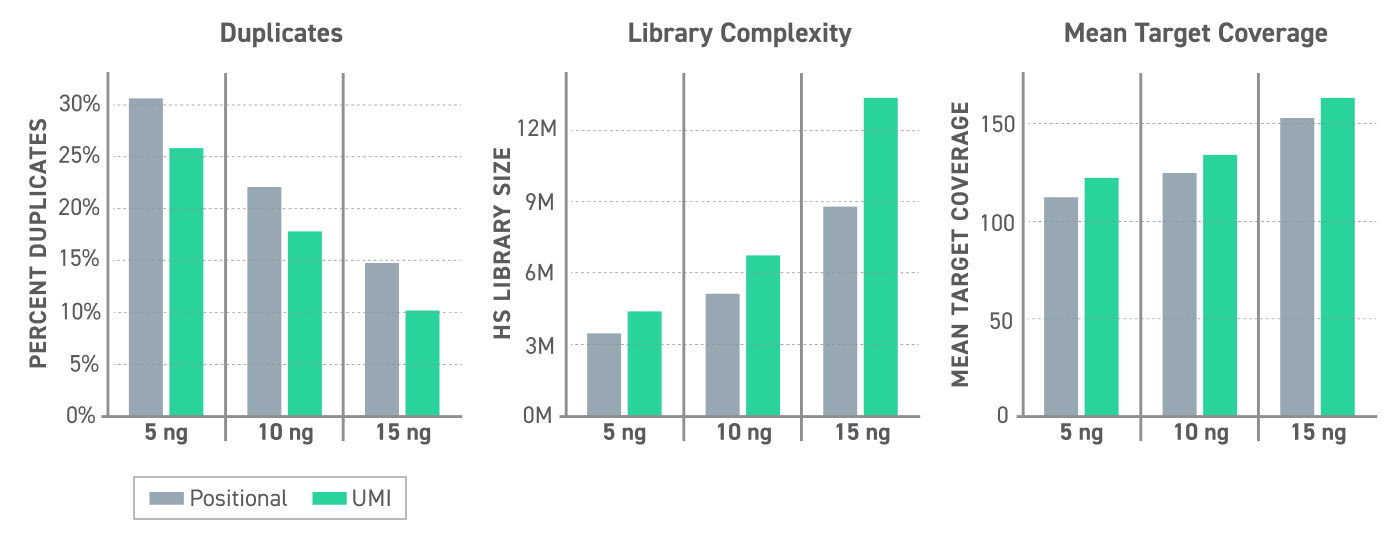
Figura 1. Rendimiento de UMI en entradas de masa de ADNlc variables. Se utilizaron 5, 10 o 15 ng de ADNlc para generar bibliotecas convertidas a EM-seq con adaptadores UMI metilados Twist después de la preparación de la biblioteca EM-Seq de NEBNext. Las bibliotecas se capturaron con el Panel de metilación Alliance Pan-cancer de Twist - 1,5 Mb utilizando el protocolo de secuenciación de metilación dirigida y se secuenciaron en un Illumina Nextseq 550 con una cobertura bruta de 1000X. Los duplicados se llamaron utilizando la herramienta GATK MarkDuplicates (método posicional estándar) o se llamaron utilizando UMIs con la herramienta GATK UmiAwareMarkDuplicatesWithMateCigar.
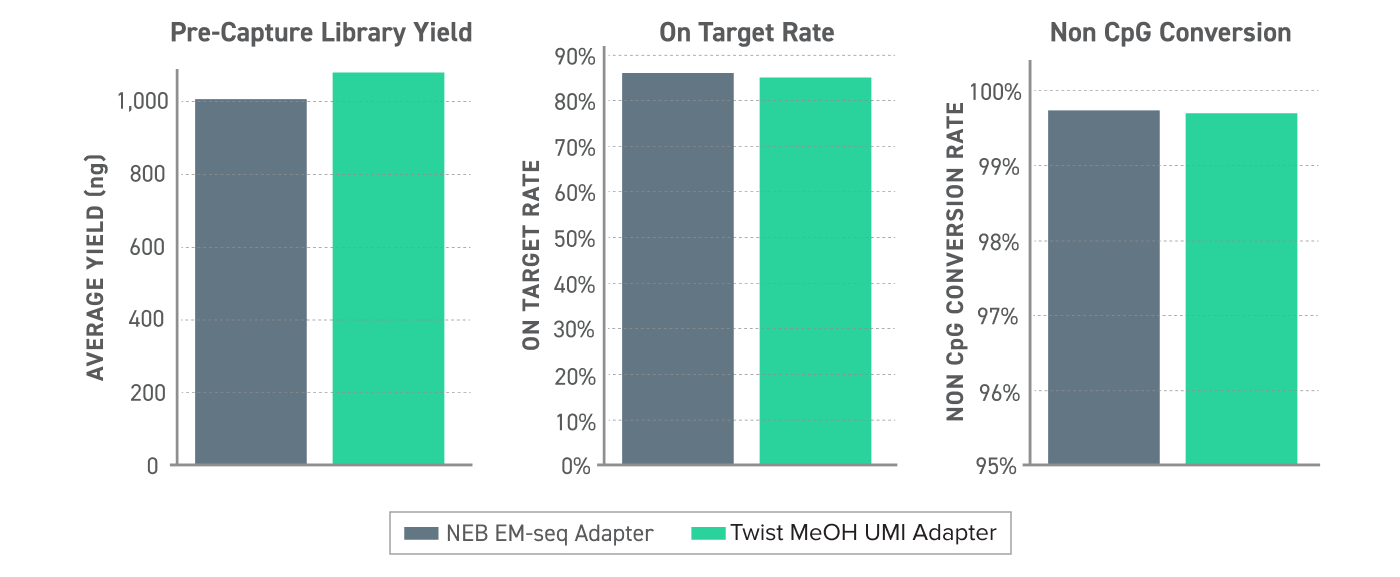
Figura 2. Rendimiento de UMI metilado en el flujo de trabajo de detección de metilación Twist 10 ng de ADNlc humano se utilizó para generar bibliotecas convertidas a EM-seq con adaptadores NEB EMseq o los nuevos adaptadores Twist Methylated UMI. Las bibliotecas se capturaron con el Panel de metilación Alliance Pan-cancer de Twist -1,5 1,5 Mb utilizando el protocolo de secuenciación de metilación dirigida y se secuenciaron en un Illumina Nextseq 550 con una cobertura bruta de 1000X.
Ejemplo de flujo de trabajo de bioinformática
La información de UMI se puede integrar en un proceso de bioinformática para eliminar duplicados antes del análisis posterior.

Figura 3. Resumen del flujo de trabajo de análisis para procesar UMI para marcado de duplicados
Las lecturas sin procesar se procesan para marcar secuencias adaptadoras y extraer información de UMI. A continuación, las lecturas se alinean con un genoma de referencia utilizando BWA-meth y se marcan con una marca duplicados utilizando la información de UMI. Después de marcar duplicados, se pueden recopilar métricas de Picard utilizando GATK y se puede realizar un análisis adicional.
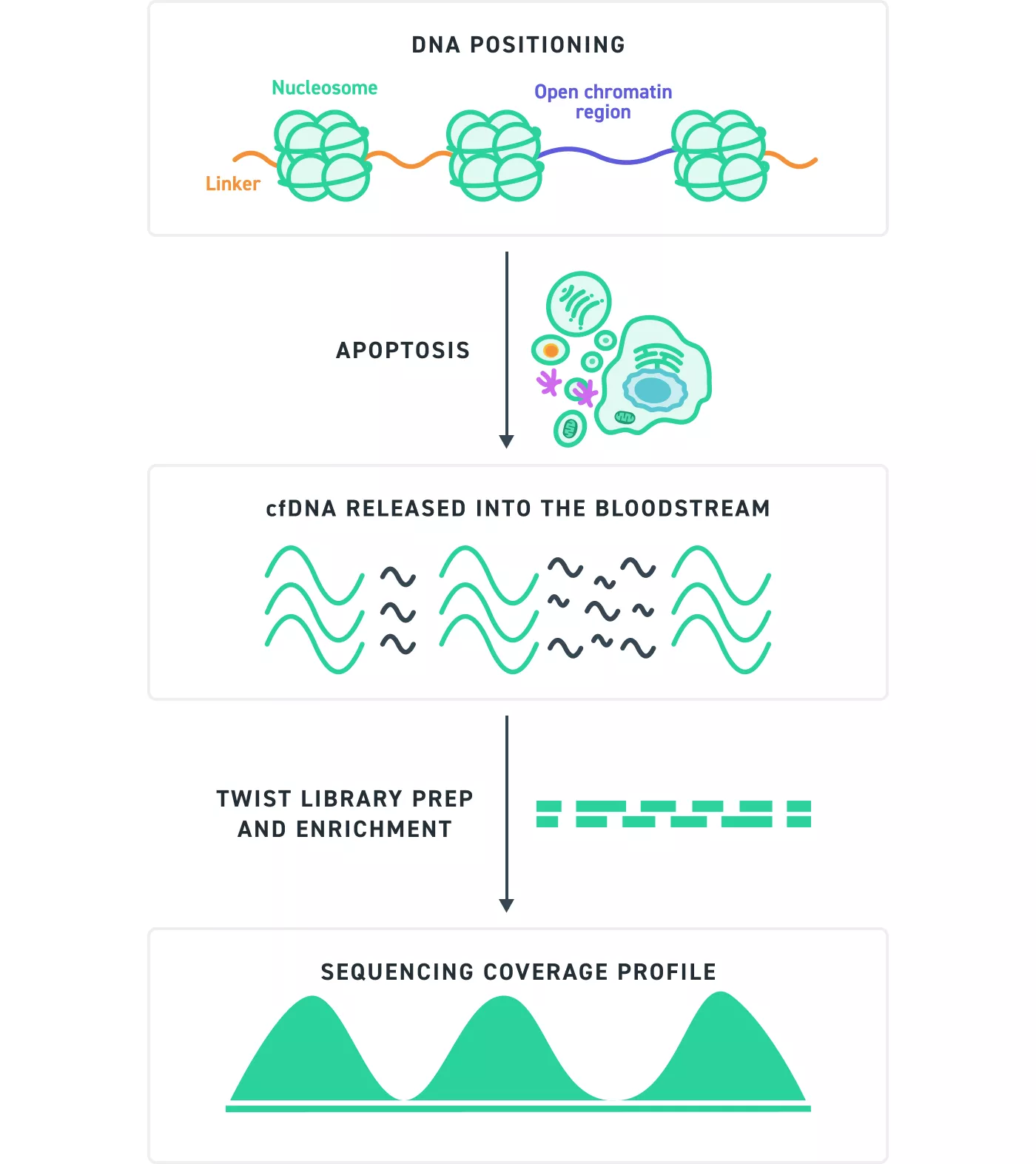
Figura 4. Esquema de la fragmentación apoptótica del ADNlc hasta la cobertura de secuenciación:
Durante la apoptosis, el ADN libre de células (ADNlc) se libera en el torrente sanguíneo. Si bien las endonucleasas fragmentan el ADN de forma aleatoria, la forma en que el ADN se envuelve alrededor de los nucleosomas introduce un sesgo posicional, lo que genera una diversidad limitada en los sitios de inicio y finalización de los fragmentos. Como resultado, moléculas distintas pueden aparecer como duplicadas en los datos de secuenciación. Los identificadores moleculares únicos (UMI) se agregan durante la preparación de bibliotecas para etiquetar moléculas individuales, lo que permite una diferenciación precisa entre duplicados verdaderos y moléculas únicas separadas.
Incorporación de UMI en el análisis para mejorar la resolución de duplicados
La llamada duplicada informada por UMI reduce los duplicados de la biblioteca en más de un 15 %, lo que genera un aumento en la complejidad de la biblioteca (25-35 %) y la cobertura media del objetivo (6-8 %) en comparación con la llamada duplicada basada en posición estándar. (Figura 1)
Figura 1. Rendimiento de UMI en entradas de masa de ADNlc variables. Se utilizaron 5, 10 o 15 ng de ADNlc para generar bibliotecas convertidas a EM-seq con adaptadores UMI metilados Twist después de la preparación de la biblioteca EM-Seq de NEBNext. Las bibliotecas se capturaron con el Panel de metilación Alliance Pan-cancer de Twist - 1,5 Mb utilizando el protocolo de secuenciación de metilación dirigida y se secuenciaron en un Illumina Nextseq 550 con una cobertura bruta de 1000X. Los duplicados se llamaron utilizando la herramienta GATK MarkDuplicates (método posicional estándar) o se llamaron utilizando UMIs con la herramienta GATK UmiAwareMarkDuplicatesWithMateCigar.
Figura 2. Rendimiento de UMI metilado en el flujo de trabajo de detección de metilación Twist 10 ng de ADNlc humano se utilizó para generar bibliotecas convertidas a EM-seq con adaptadores NEB EMseq o los nuevos adaptadores Twist Methylated UMI. Las bibliotecas se capturaron con el Panel de metilación Alliance Pan-cancer de Twist -1,5 1,5 Mb utilizando el protocolo de secuenciación de metilación dirigida y se secuenciaron en un Illumina Nextseq 550 con una cobertura bruta de 1000X.
Ejemplo de flujo de trabajo de bioinformática
La información de UMI se puede integrar en un proceso de bioinformática para eliminar duplicados antes del análisis posterior.
Figura 3. Resumen del flujo de trabajo de análisis para procesar UMI para marcado de duplicados
Las lecturas sin procesar se procesan para marcar secuencias adaptadoras y extraer información de UMI. A continuación, las lecturas se alinean con un genoma de referencia utilizando BWA-meth y se marcan con una marca duplicados utilizando la información de UMI. Después de marcar duplicados, se pueden recopilar métricas de Picard utilizando GATK y se puede realizar un análisis adicional.
Figura 4. Esquema de la fragmentación apoptótica del ADNlc hasta la cobertura de secuenciación:
Durante la apoptosis, el ADN libre de células (ADNlc) se libera en el torrente sanguíneo. Si bien las endonucleasas fragmentan el ADN de forma aleatoria, la forma en que el ADN se envuelve alrededor de los nucleosomas introduce un sesgo posicional, lo que genera una diversidad limitada en los sitios de inicio y finalización de los fragmentos. Como resultado, moléculas distintas pueden aparecer como duplicadas en los datos de secuenciación. Los identificadores moleculares únicos (UMI) se agregan durante la preparación de bibliotecas para etiquetar moléculas individuales, lo que permite una diferenciación precisa entre duplicados verdaderos y moléculas únicas separadas.
110830
Producto: Sistema de adaptadores de UMI metilados de Twist compatible con TruSeq, 96 muestra110830
Producto: Sistema de adaptadores de UMI metilados de Twist compatible con TruSeq, 96 muestra Ficha del producto
Ficha del producto

