Exome Sequencing 101: Part 2 - Target Enrichment
Welcome to Part 2 of Exome Sequencing 101. After sample and library preparation, the next step in the exome sequencing workflow is target enrichment. Target enrichment is the process of isolating and separating relevant regions of the genome for focused analysis by next generation sequencing (NGS).
Why would you perform target enrichment? This step is crucial in workflows that require labor-intensive parallel sequencing, routines that deliver clinical results, or research into specific mutations. Once a researcher has a target enrichment library for a set of genes, they can run sequencing analyses more efficiently by increasing sequencing throughput, or provide results to patients more quickly. 80% of inherited diseases are caused by mutations within 1% of the genome, so sequencing targeted portions of the genome even makes sense when no particular site is under inspection for mutation. Ultimately, target enrichment is about decluttering your workspace so you can get more work done.
Let’s continue following the common clinical scenario our last piece introduced:
A hospital staff has fully prepared a DNA library from a patient with an unknown inherited disease. The doctors studying the patient’s case are under pressure to provide the patient with treatment options as soon as possible. To work efficiently, they need to be able to identify regions of the genome that are likely sites of disease mutations.
As we saw in our last piece (Exome Sequencing 101: Part 1 - Library Preparation), a genomic DNA library has been prepared, providing the researcher with access to a sample of the patient’s genome. Having the genome available for study may be useful in exploratory research scenarios, but when a specific region of the genome is the focus of study, or when a subset of the genome that may contain disease-causing mutations is under investigation, having to manage a whole genome can be unnecessary, cost-prohibitive, and resource-intensive.
What’s the target enrichment process?
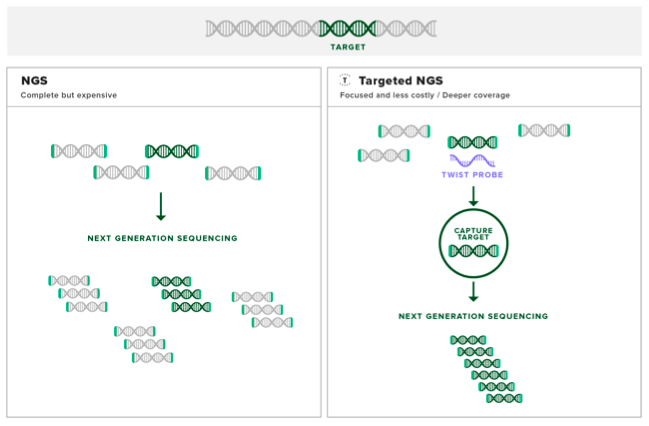
Hybridization-based target enrichment is attuned to research dealing with large genomic regions of interest, such as pan-cancer panels, or the protein-coding exome.
Stepwise approach to hybridization-based target enrichment
Step One: design and acquire probes. Probes are small strands of either single-stranded DNA, single-stranded RNA or double-stranded DNA that are designed to contain specific sequences that are complementary to the genomic region of interest. Probes can vary in length between about 50 to 120 bp.
In the clinical example, our hypothetical patient is undergoing a common type of target enrichment called exome sequencing. For example, Twist Bioscience’s Core Human Exome Enrichment Kit contains double-stranded DNA probes designed to bind complementary to the exome DNA sequences in the patient’s genomic sample associated with all known inherited disease.
Step Two: use the probes to extract target DNA. The probes are mixed with the genomic sample and then heated to above 95°C to melt the base pair interactions in the double-stranded genomic DNA, forming a pool of single stranded DNA. Bringing the temperature down allows the genomic DNA to start to form back into complementary double stranded molecules. As the probes are designed to be complementary with the exome, they will also form base pair interactions with the genomic DNA.
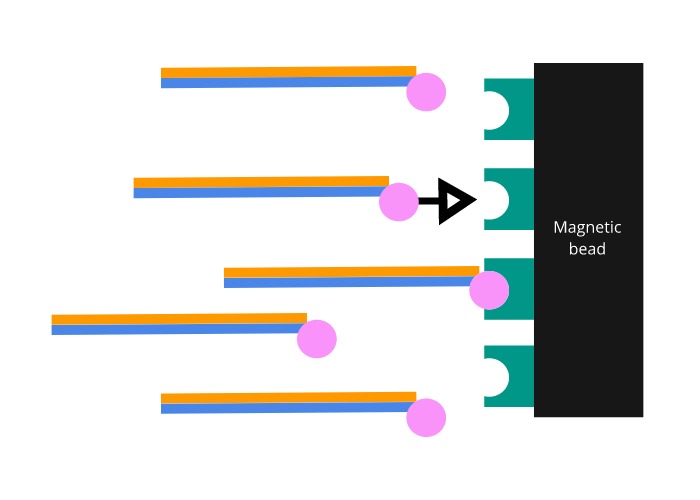
The probes are manufactured to include a biotin molecule on one end. Biotin is a molecule that binds to a protein called streptavidin with one of the strongest interactions known to biology. Magnetic beads coated in streptavidin are then added to the mixture. Once the coated beads are firmly bound to the biotinylated probes, a magnet is used to pull the bound exome DNA out of solution.
🤔 Why is target capture necessary?
With limited time and resources, researchers often have difficult decisions to make, particularly when it comes to sequencing. Researchers have to be judicious in their studies, balancing the depth, breadth, and scale of sequencing in order to get the most discovery bang for their buck. Read our recent article to learn more about how target enrichment helps you reduce costs without compromising on data quality: Capturing the Basics of NGS Target Enrichment
Choosing a target enrichment platform is an application-driven decision, where the experiment’s specific requirements, and the availability of research equipment determine the method. Hybridization capture enrichment is a complicated procedure that is simplified by commercially available kits, and made efficient by acquiring high-quality probes like those synthesized by Twist Bioscience.
Twist Bioscience’s target capture probes are unique in the enrichment market as they consist of double stranded DNA. During the melt step the probes unwind, becoming two independent probes of complementary sequence. When the genome fragments unwind, both strands are captured independently. Each genome fragment can be sequenced twice. Also, with some genomic fragments, one of the two strands may be difficult to capture due to unfavorable sequence composition. By using double stranded probes, capture efficiency can be maximised as there are multiple opportunities to capture a single fragment.
The target enrichment part of an NGS workflow can be critical for experiment efficiency. An Illumina HiSeq4000 sequencing machine is estimated to process 6 whole genomes simultaneously over 3 days, but can process 90 exomes in just 2 days. By extracting just the exome, sequencing productivity can increase by over 2,000% per week.
Data shows that uniform synthesis of probes is important for downstream productivity. Additionally, using next generation DNA synthesis technology, capture probe diversity can be finely tuned allowing for genome fragments to be captured uniformly.
For example, Twist Bioscience’s target enrichment library designs result in a 40% lower requirement for sequencing to ensure 80% of targets obtain 20 or more reads. This leads to capturing higher library diversity/complexity, or unique molecules, which leads to increased confidence in variant calling. This also engenders fewer gigabytes of sequencing required to achieve equivalent read depth, which ultimately dramatically reduces sequencing cost per exome. With smaller sequence pools, bioinformatics resources are also freed up to analyze more samples, or screen multiple patients simultaneously.
For our example patient mentioned earlier, this means both cost and wait time for exomic screening is dramatically reduced over genomic screening. With the vast majority of disease-causing mutations occuring in a small fraction of the genome, why screen the entire genome when you don’t have to?
いかがでしたか?
気に入った
気に入らなかった
大好き
驚かされた
面白かった

