Verbesserter Methylierungsnachweis für Flüssigbiopsie-Workflows
Flüssigbiopsie-Technologien sind dabei, die Landschaft der Krebsfrüherkennung und -überwachung zu verändern. Flüssigbiopsien bieten einen minimalinvasiven Ansatz und ermöglichen die Analyse von zirkulierenden Biomarkern wie cfDNA (zellfreie DNA), die wertvolle Informationen über krebsassoziierte genetische und epigenetische Veränderungen enthalten. Unter diesen Biomarkern sind DNA-Methylierungsänderungen ein vielversprechender Weg, um verschiedene Krebsarten mit hoher Spezifität und Sensitivität zu identifizieren und zu verfolgen.
Trotz ihres Potenzials stellt die Arbeit mit cfDNA aufgrund ihrer begrenzten Menge und ihrer ausgeprägten Fragmentierungsmuster eine besondere Herausforderung dar. Die meisten cfDNA-Fragmente sind etwa 167 bp lang, was auf ihren nukleosomgebundenen Ursprung hinweist. Diese Fragmentierung führt zu:
- Geringer Stopp-Start-Diversität, was zu mehr einzigartigen Molekülen führt, die als doppelte Sequenzierungs-Reads bezeichnet werden
- Geringerer Library-Komplexität, was die Empfindlichkeit des Assays beeinträchtigen kann
Diese inhärenten Eigenschaften machen herkömmliche Methylierungsnachweisworkflows für die cfDNA-Analyse weniger effektiv, so dass fortschrittliche Tools und Techniken erforderlich sind, die auf diese Einschränkungen zugeschnitten sind.
Die Twist Methylated UMI Adapter verbessern cfDNA-Methylierungsworkflows, indem sie eine genaue Deduplizierung durch eindeutige molekulare Identifikatoren (Unique Molecular Identifiers, UMIs) ermöglichen und so falsche Duplikataufrufe in Proben mit geringer Diversität reduzieren. Ihr Design gewährleistet die Kompatibilität mit EM-Seq-Protokollen, bewahrt die Fragmentlänge, maximiert die verwertbaren Daten und führt zu einer verbesserten Target-Abdeckung und Reproduzierbarkeit, wodurch sie zu einem wichtigen Instrument für hochzuverlässige Methylierungsstudien werden.

Verbesserter Methylierungsnachweis für Flüssigbiopsie-Workflows
Flüssigbiopsie-Technologien sind dabei, die Landschaft der Krebsfrüherkennung und -überwachung zu verändern. Flüssigbiopsien bieten einen minimalinvasiven Ansatz und ermöglichen die Analyse von zirkulierenden Biomarkern wie cfDNA (zellfreie DNA), die wertvolle Informationen über krebsassoziierte genetische und epigenetische Veränderungen enthalten. Unter diesen Biomarkern sind DNA-Methylierungsänderungen ein vielversprechender Weg, um verschiedene Krebsarten mit hoher Spezifität und Sensitivität zu identifizieren und zu verfolgen.
Trotz ihres Potenzials stellt die Arbeit mit cfDNA aufgrund ihrer begrenzten Menge und ihrer ausgeprägten Fragmentierungsmuster eine besondere Herausforderung dar. Die meisten cfDNA-Fragmente sind etwa 167 bp lang, was auf ihren nukleosomgebundenen Ursprung hinweist. Diese Fragmentierung führt zu:
- Geringer Stopp-Start-Diversität, was zu mehr einzigartigen Molekülen führt, die als doppelte Sequenzierungs-Reads bezeichnet werden
- Geringerer Library-Komplexität, was die Empfindlichkeit des Assays beeinträchtigen kann
Diese inhärenten Eigenschaften machen herkömmliche Methylierungsnachweisworkflows für die cfDNA-Analyse weniger effektiv, so dass fortschrittliche Tools und Techniken erforderlich sind, die auf diese Einschränkungen zugeschnitten sind.
Die Twist Methylated UMI Adapter verbessern cfDNA-Methylierungsworkflows, indem sie eine genaue Deduplizierung durch eindeutige molekulare Identifikatoren (Unique Molecular Identifiers, UMIs) ermöglichen und so falsche Duplikataufrufe in Proben mit geringer Diversität reduzieren. Ihr Design gewährleistet die Kompatibilität mit EM-Seq-Protokollen, bewahrt die Fragmentlänge, maximiert die verwertbaren Daten und führt zu einer verbesserten Target-Abdeckung und Reproduzierbarkeit, wodurch sie zu einem wichtigen Instrument für hochzuverlässige Methylierungsstudien werden.
Einbeziehung von UMIs in die Analyse zur verbesserten Auflösung von Duplikaten
Durch Duplikataufrufe mithilfe von UMIs werden Library-Duplikate um mehr als 15 % reduziert, was zu einem Anstieg der Library-Komplexität (25–35 %) und der mittleren Target-Abdeckung (6–8 %) im Vergleich zu standardmäßigen positionsbasierten Duplikataufrufen führt. (Abbildung 1)
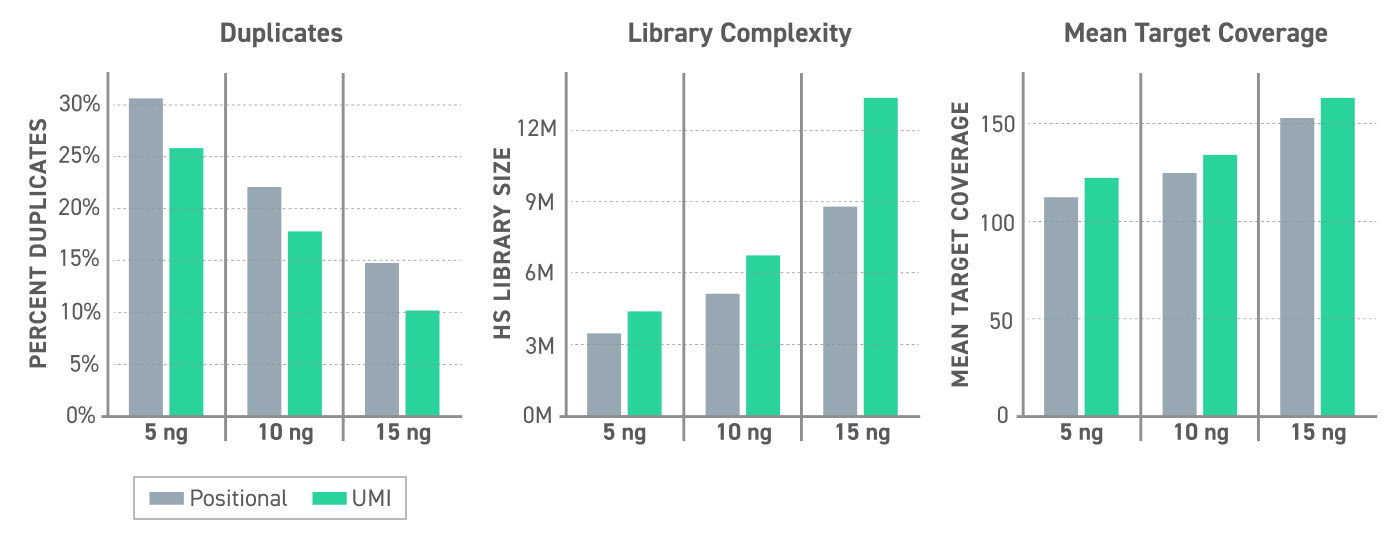
Abbildung 1. UMI-Leistung bei variablen cfDNA-Masseninputs 5, 10 oder 15 ng cfDNA wurden verwendet, um EM-seq-konvertierte Librarys mit Twist Methylated UMI Adaptern nach der NEBNext EM-Seq-Libraryvorbereitung zu erzeugen. Die Librarys wurden mit dem Twist Alliance Pan-cancer Methylation Panel – 1,5 MB unter Verwendung des Targeted Methylation Sequencing Protocol erfasst und auf einem Illumina Nextseq 550 auf eine 1.000-fache Rohabdeckung sequenziert. Duplikate wurden mit dem GATK MarkDuplicates-Tool (Standard-Positionsmethode) oder mit UMIs mit dem GATK UmiAwareMarkDuplicatesWithMateCigar-Tool aufgerufen.
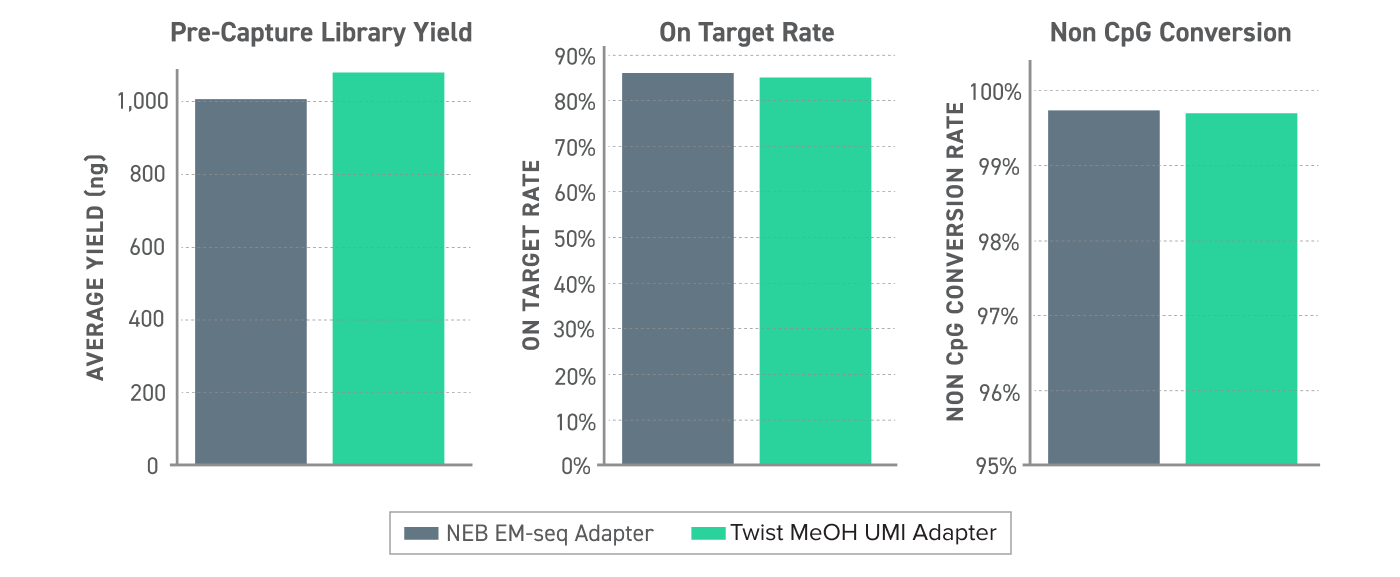
Abbildung 2. Methylierte UMI-Leistung im Twist Methylation Detection Workflow 10 ng humaner cfDNA wurden verwendet, um EM-seq-konvertierte Librarys mit NEB EMseq-Adaptern oder den neuen Twist Methylated UMI Adaptern zu erzeugen. Die Librarys wurden mit dem Twist Alliance Pan-cancer Methylation Panel – 1,5 MB unter Verwendung des Targeted Methylation Sequencing Protocol erfasst und auf einem Illumina Nextseq 550 auf eine 1.000-fache Rohabdeckung sequenziert.
Beispiel für einen Bioinformatik-Workflow
UMI-Informationen können in eine Bioinformatik-Pipeline integriert werden, um Duplikate vor der nachgeschalteten Analyse zu entfernen.

Abbildung 3. Zusammenfassung des Analyse-Workflows zur Verarbeitung von UMIs für die Duplikatkennzeichnung
Roh-Reads werden verarbeitet, um Adaptersequenzen zu kennzeichnen und UMI-Informationen zu extrahieren. Die Reads werden dann mit BWA-meth an einem Referenzgenom ausgerichtet und Duplikate werden mit UMI-Informationen markiert. Nach der Duplikatkennzeichnung können Picard-Metriken mit GATK erfasst und zusätzliche Analysen durchgeführt werden.
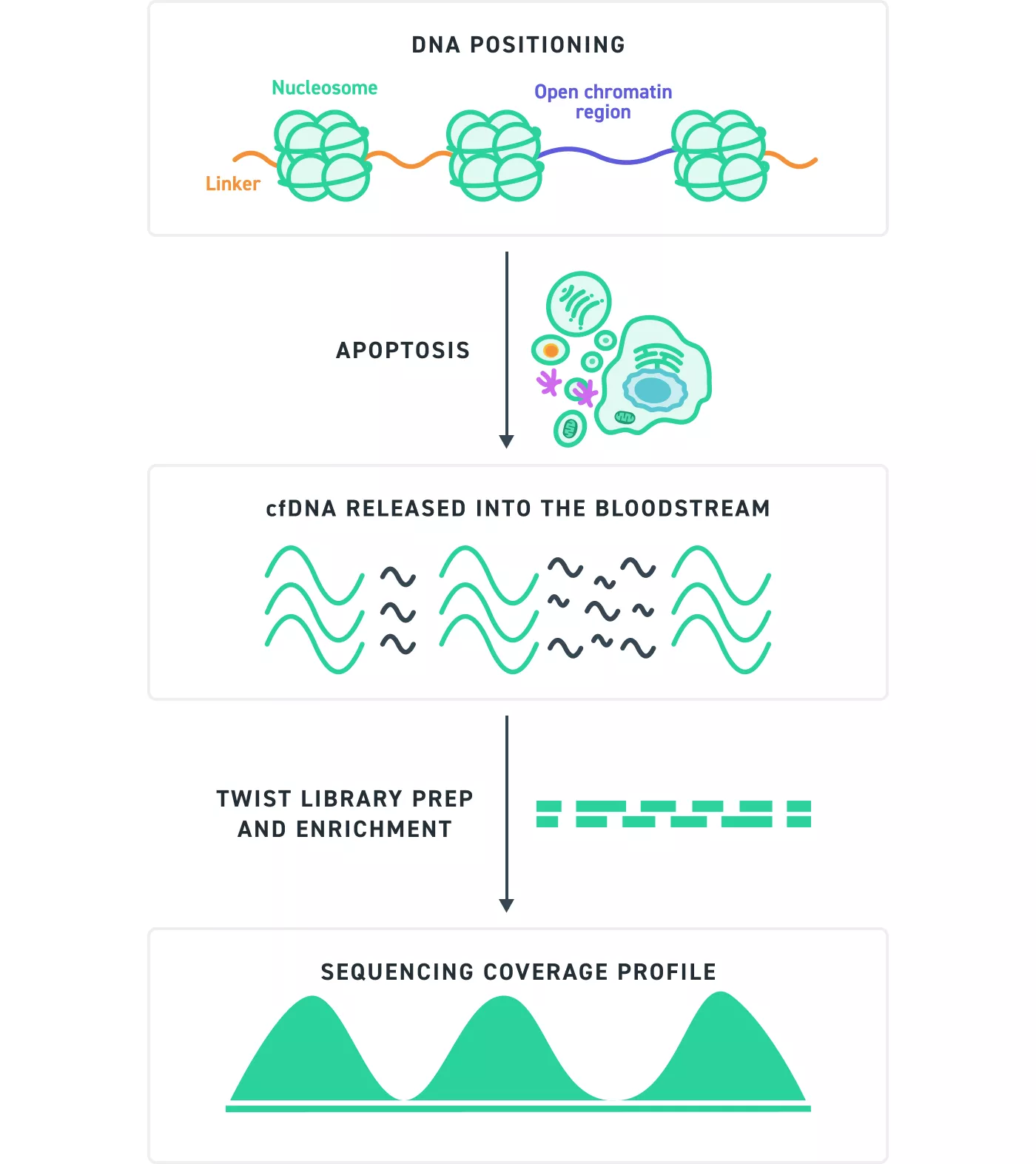
Abbildung 4. Schematische Darstellung der apoptotischen cfDNA-Fragmentierung zur Sequenzierungsabdeckung:
Während der Apoptose wird zellfreie DNA (cfDNA) in den Blutkreislauf freigesetzt. Während Endonukleasen die DNA zufällig fragmentieren, entsteht durch die Art und Weise, wie die DNA um Nukleosomen gewickelt ist, ein Positionsfehler, der zu einer eingeschränkten Diversität der Start- und Stoppstellen der Fragmente führt. Dies hat zur Folge, dass unterschiedliche Moleküle in den Sequenzierungsdaten möglicherweise als Duplikate erscheinen. Während der Libraryvorbereitung werden eindeutige molekulare Identifikatoren (UMI) hinzugefügt, um einzelne Moleküle zu kennzeichnen. Dies ermöglicht eine genaue Unterscheidung zwischen echten Duplikaten und separaten, eindeutigen Molekülen.
Einbeziehung von UMIs in die Analyse zur verbesserten Auflösung von Duplikaten
Durch Duplikataufrufe mithilfe von UMIs werden Library-Duplikate um mehr als 15 % reduziert, was zu einem Anstieg der Library-Komplexität (25–35 %) und der mittleren Target-Abdeckung (6–8 %) im Vergleich zu standardmäßigen positionsbasierten Duplikataufrufen führt. (Abbildung 1)
Abbildung 1. UMI-Leistung bei variablen cfDNA-Masseninputs 5, 10 oder 15 ng cfDNA wurden verwendet, um EM-seq-konvertierte Librarys mit Twist Methylated UMI Adaptern nach der NEBNext EM-Seq-Libraryvorbereitung zu erzeugen. Die Librarys wurden mit dem Twist Alliance Pan-cancer Methylation Panel – 1,5 MB unter Verwendung des Targeted Methylation Sequencing Protocol erfasst und auf einem Illumina Nextseq 550 auf eine 1.000-fache Rohabdeckung sequenziert. Duplikate wurden mit dem GATK MarkDuplicates-Tool (Standard-Positionsmethode) oder mit UMIs mit dem GATK UmiAwareMarkDuplicatesWithMateCigar-Tool aufgerufen.
Abbildung 2. Methylierte UMI-Leistung im Twist Methylation Detection Workflow 10 ng humaner cfDNA wurden verwendet, um EM-seq-konvertierte Librarys mit NEB EMseq-Adaptern oder den neuen Twist Methylated UMI Adaptern zu erzeugen. Die Librarys wurden mit dem Twist Alliance Pan-cancer Methylation Panel – 1,5 MB unter Verwendung des Targeted Methylation Sequencing Protocol erfasst und auf einem Illumina Nextseq 550 auf eine 1.000-fache Rohabdeckung sequenziert.
Beispiel für einen Bioinformatik-Workflow
UMI-Informationen können in eine Bioinformatik-Pipeline integriert werden, um Duplikate vor der nachgeschalteten Analyse zu entfernen.
Abbildung 3. Zusammenfassung des Analyse-Workflows zur Verarbeitung von UMIs für die Duplikatkennzeichnung
Roh-Reads werden verarbeitet, um Adaptersequenzen zu kennzeichnen und UMI-Informationen zu extrahieren. Die Reads werden dann mit BWA-meth an einem Referenzgenom ausgerichtet und Duplikate werden mit UMI-Informationen markiert. Nach der Duplikatkennzeichnung können Picard-Metriken mit GATK erfasst und zusätzliche Analysen durchgeführt werden.
Abbildung 4. Schematische Darstellung der apoptotischen cfDNA-Fragmentierung zur Sequenzierungsabdeckung:
Während der Apoptose wird zellfreie DNA (cfDNA) in den Blutkreislauf freigesetzt. Während Endonukleasen die DNA zufällig fragmentieren, entsteht durch die Art und Weise, wie die DNA um Nukleosomen gewickelt ist, ein Positionsfehler, der zu einer eingeschränkten Diversität der Start- und Stoppstellen der Fragmente führt. Dies hat zur Folge, dass unterschiedliche Moleküle in den Sequenzierungsdaten möglicherweise als Duplikate erscheinen. Während der Libraryvorbereitung werden eindeutige molekulare Identifikatoren (UMI) hinzugefügt, um einzelne Moleküle zu kennzeichnen. Dies ermöglicht eine genaue Unterscheidung zwischen echten Duplikaten und separaten, eindeutigen Molekülen.
110830
Produkt: Twist Methylated UMI Adapters – TruSeq-kompatibel, 96 Proben110830
Produkt: Twist Methylated UMI Adapters – TruSeq-kompatibel, 96 Proben Produktblatt
Produktblatt

