Proteinstrukturvorhersage: Neue Werkzeuge, um diese komplexen Lebensstrukturen zu verstehen
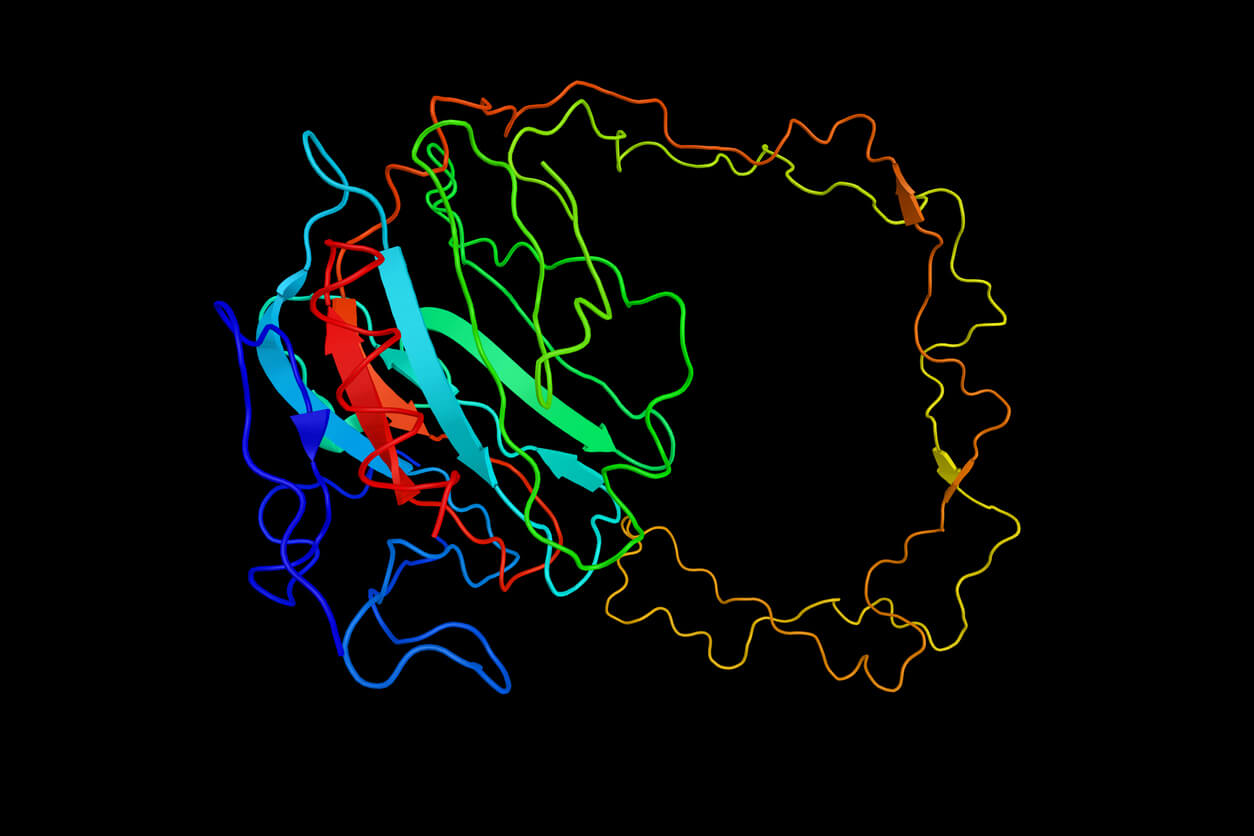
Proteine sind die molekularen Maschinen des Lebens. Sie beginnen als Ketten von Aminosäuren, die aus einer entsprechenden RNA-Sequenz translatiert werden, und erhalten ihre Funktionalität durch Faltung in komplexe 3D-Strukturen. Die Natur lässt den Prozess einfach erscheinen. Obwohl es Millionen möglicher 3D-Konfigurationen gibt, falten sich die meisten kleinen Proteine durch verschiedene atomare Kräfte augenblicklich zu einer stabilen funktionellen Struktur. Das Verständnis, wie sich Proteine falten, ist der Schlüssel zur Beantwortung vieler Fragen in der Biologie. Wie führen beispielsweise Mutationen im Genom zu Krankheiten oder wie kann ein Enzym gentechnisch so verändert werden, dass Kunststoff effektiv abgebaut wird?
Die ultimative Frage – das mittlerweile 50 Jahre alte Postulat „Das Problem der Proteinfaltung“ – lautet, ob die Struktur eines Proteins allein anhand seiner Aminosäuresequenz vorhergesagt werden kann. Bisher wurde die Frage mit einem definitiven Nein beantwortet. Bei einer Aminosäurekette gibt es einfach zu viele mögliche Strukturen, um sie zu kennen, das ist richtig.
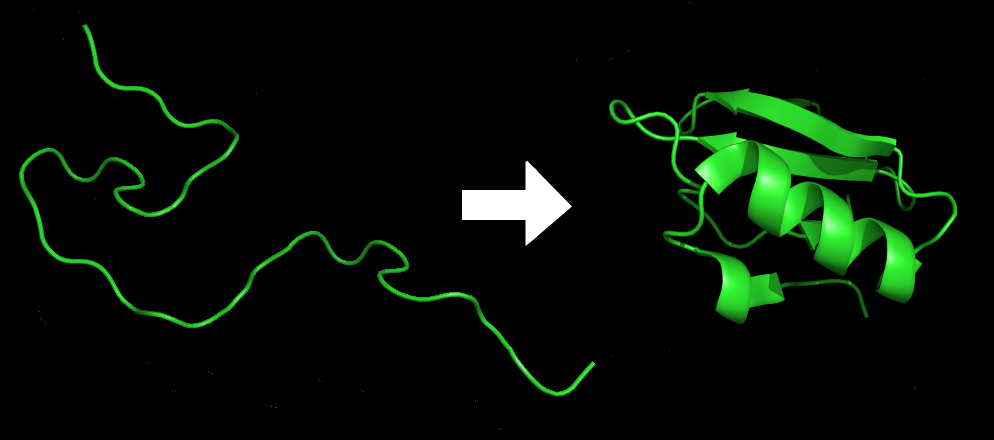
An amino acid chain for Chymotrypsin inhibitor 2 (1LW6) in its unfolded and folded form. Source: Wikicommons.
Typischerweise werden Proteinstrukturen durch Beobachtung des Beugungsmusters von Röntgenstrahlen bestimmt, die durch ihre kristallisierten Formen geleitet werden. Die Röntgenbestrahlung von Proteinkristallen ist einfach, aber das Erstellen dieser Kristalle ist schwierig. Viele bezeichnen es als „schwarze Kunst“, da die Identifizierung der richtigen Bedingungen für die Keimbildung eines Proteinkristalls erneut eine Herausforderung darstellt und typischerweise durch rohe Gewalt oder reines Glück erreicht wird. In der Literatur gibt es sogar Beispiele für einzelne Wimpern, die ein Experiment kontaminieren, aber den perfekten Keim für die Kristallkeimbildung abgeben. Alternative Methoden wie Kryo-Elektronenmikroskopie und Kernspinresonanzspektroskopie wurden ebenfalls entwickelt, um Proteinstrukturen aufzulösen. Alle diese laborbasierten Techniken sind jedoch sowohl teuer als auch zeitaufwändig.
Aus diesem Grund haben Wissenschaftler versucht, Berechnungsmethoden zur Lösung von Proteinstrukturen zu entwickeln, die ausschließlich auf ihrer Aminosäuresequenz basieren. Trotz erheblicher Anstrengungen ist das Gebiet seit fast 50 Jahren von kleinen Fortschritten bei diesen Methoden geprägt – bis jetzt.
CASP-Wettbewerb gibt Gewinner bekannt
Alle zwei Jahre findet eine Konferenz zur Beurteilung der neuesten Entwicklungen bei der rechnergestützten Auflösung von Proteinstrukturen statt. Bei dieser alle zwei Jahre stattfindenden CASP-Konferenz (Critical Assessment of Techniques for Protein Structure Prediction) werden Teams aufgefordert, erst kürzlich mithilfe von Labormethoden nachgewiesene Proteinstrukturen rechnerisch aufzulösen.
At this year’s iteration, CASP14, Google’s DeepMind blew the competition away with its artificial intelligence platform AlphaFold 2. Not only did it handily outperform its competitors, it did as well as lab-based techniques for over two-thirds of the protein structures it was given to solve.
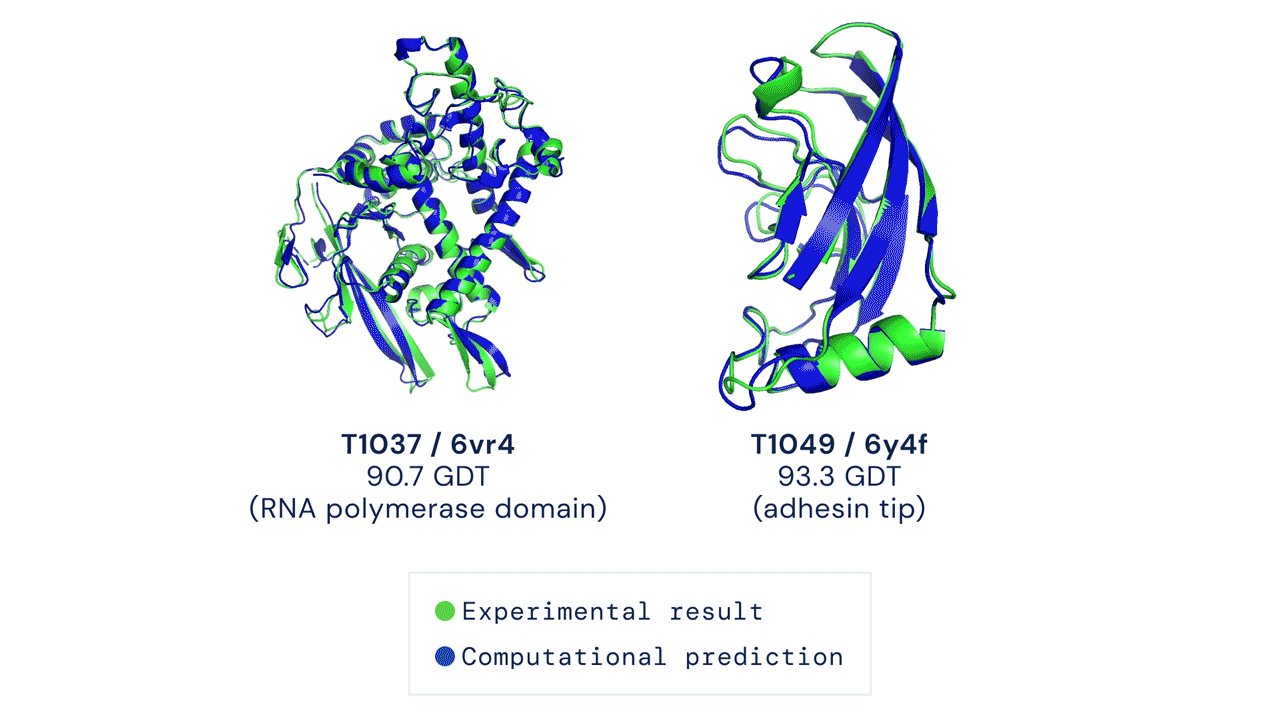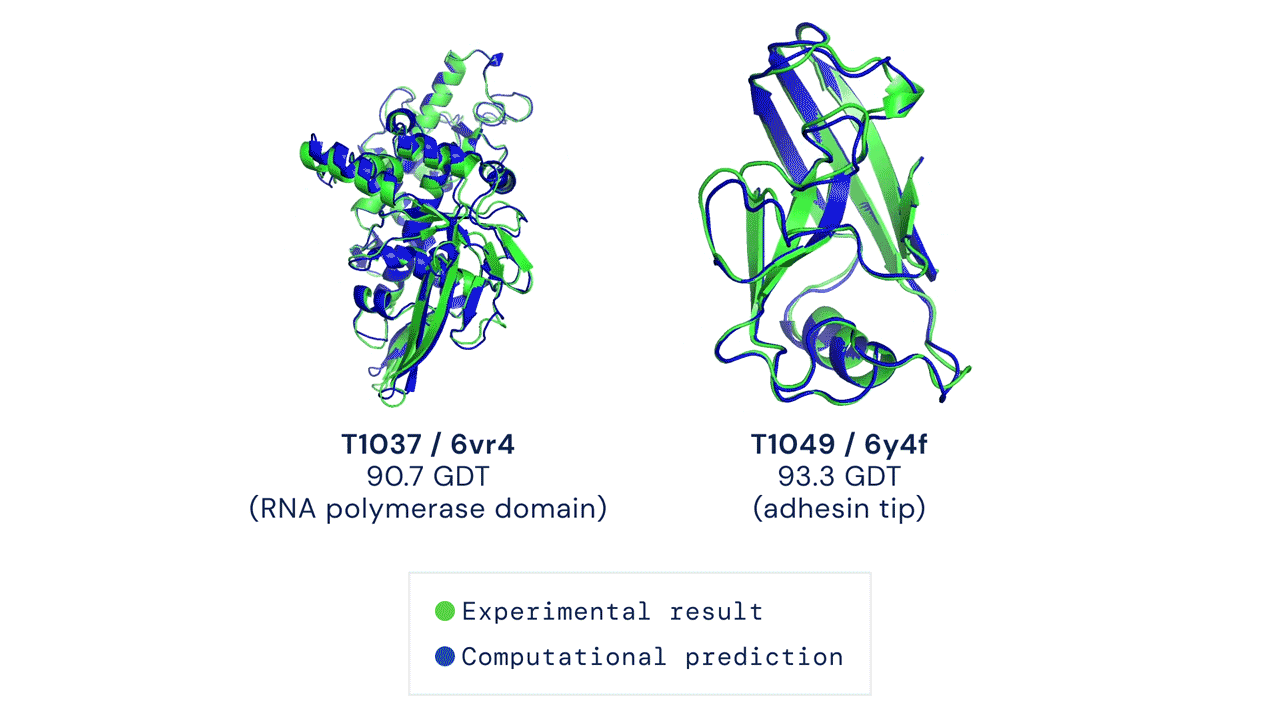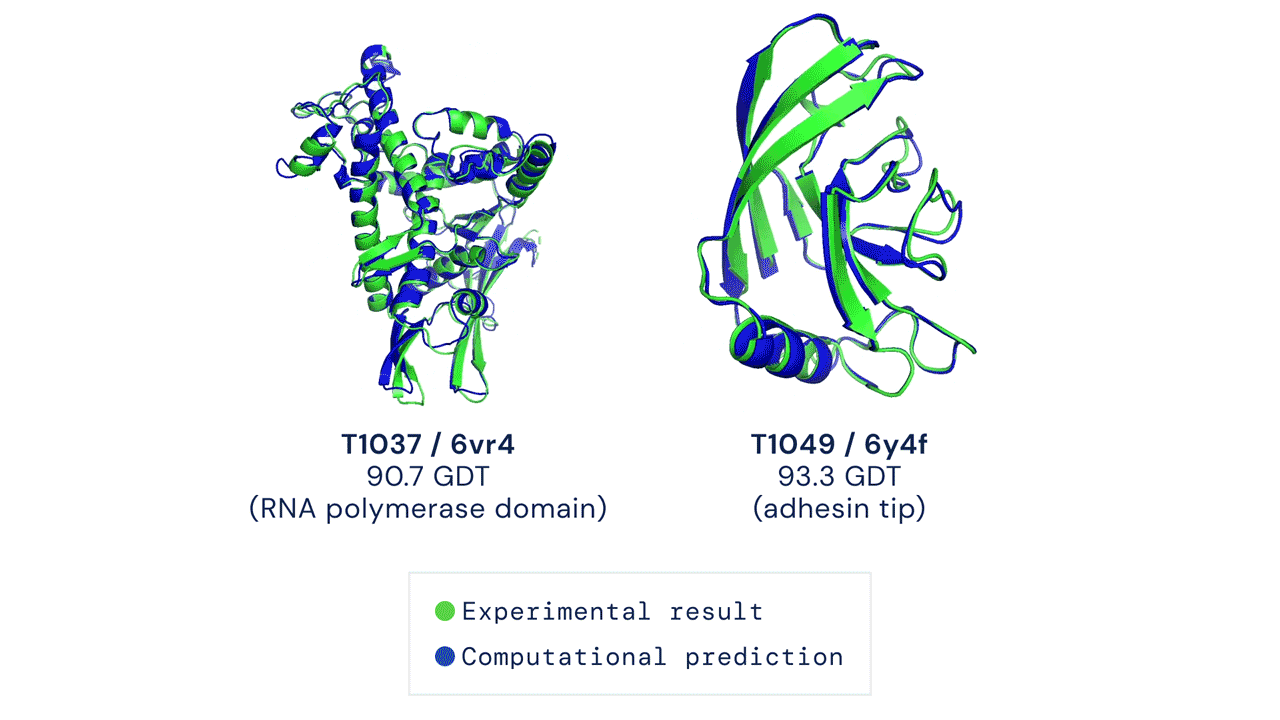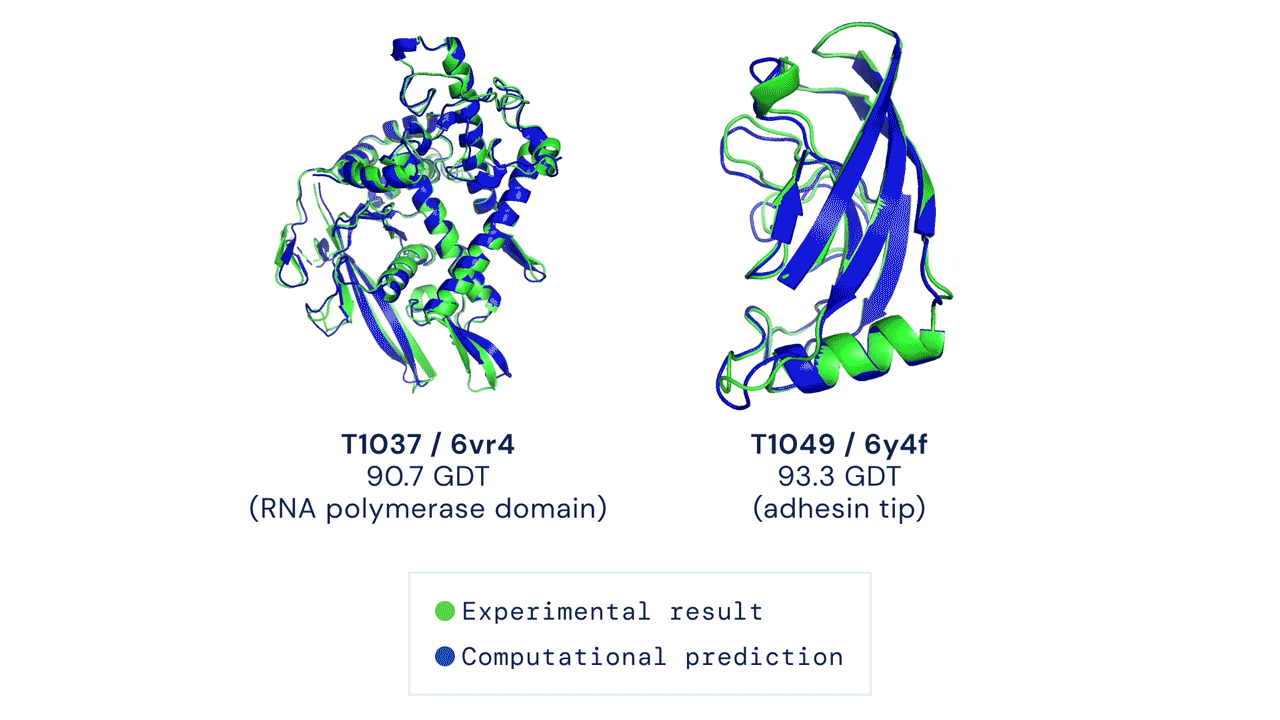
Zwei von AlphaFold vorhergesagte Proteinstrukturen überlagerten ihre experimentell abgeleiteten Strukturen. Quelle
CASP misst die Genauigkeit von Proteinfaltungsalgorithmen mit einer Metrik, die als Global Distance Test (GDT) bezeichnet wird. Im Bereich von 0 bis 100 bewertet der GDT im Wesentlichen, wie nahe eine rechnerische Vorhersage an der im Labor ermittelten Struktur liegt. AlphaFold 2 erzielte beim CASP14 einen GDT-Medianwert von 92,4. Ein solch hohes Ergebnis macht es schwierig festzustellen, ob die rechnergestützt oder die empirisch gewonnenen Strukturdaten „korrekter“ sind.
Under the Hood of AlphaFold 2
The AlphaFold algorithm uses deep learning to solve protein structures. Deep learning is a form of machine learning that performs a task by automatically extracting the features of a system (e.g. 3D structure) from raw input data (e.g. many protein structures and amino acid chains). Although the inner workings of AlphaFold 2 won’t be published until early 2021, what’s clear is that it uses a deep-learning technique called an attention network.
Ein ähnlicher Ansatz wird verwendet, um Maschinen das Verstehen von Sprache beizubringen. Stellen Sie sich diesen Blog-Artikel als eine lange Aminosäurekette vor. Genau wie die Aminosäurekette durch eine Reihe lokaler und entfernter Wechselwirkungen zwischen Aminosäuren komplexe Proteinstrukturen bildet, vermittelt dieser Artikel Bedeutung durch lokale und entfernte Beziehungen zwischen Wörtern, Sätzen und Absätzen. Attention-basierte Deep Learning-Algorithmen synthetisieren diese lokalen und entfernten Beziehungen, um Aufgaben wie das Verstehen der Bedeutung von Sprache oder das Bestimmen der 3D-Struktur eines Proteins zu erlernen.
Weniger abstrakt ausgedrückt: AlphaFold 2 kombinierte Informationen, die aus evolutionär verwandten Sequenzen, multiplen Sequenzalignments und Wechselwirkungen zwischen Aminosäurerestpaaren abgeleitet wurden, um zu erfahren, wie diese Variablen innerhalb der 170.000 bekannten Proteinstrukturen, die zum Trainieren des Algorithmus verwendet werden, zusammenhängen.
Kann AlphaFold 2 reale Probleme lösen?
Die Fähigkeit der Plattform, Strukturen in Wochen statt in Monaten (oder Jahren) aufzulösen, weist auf eine Rolle der KI-Plattform bei der therapeutischen Entdeckung kleiner Moleküle und der Entwicklung neuer Proteine für eine Vielzahl realer Anwendungen hin – von der Optimierung industrieller Prozesse bis zum Abbau von Kunststoff.
Computational methods are frequently used to determine how drugs interact with their protein targets. Unfortunately, the structure of thousands of druggable proteins remains unknown, making it difficult to develop effective drugs against them. Predictive algorithms like AlphaFold 2 are promising solutions to this backlog if shown to be consistently accurate. In a proof-of-principle demonstration, AlphaFold 2 accurately predicted the structure of the SARS-CoV-2 Spike protein, the main therapeutic target against COVID-19, earlier this year. It also predicted several other SARS-CoV-2 proteins before they were confirmed by lab-based methods.
Another game-changing application where AlphaFold 2 may make its mark is in protein engineering. Often called the inverse protein-folding problem, protein design aims to identify amino acid sequences that will form stabilized, functional, useful protein structures. It encapsulates everything from engineering receptors with different ligand specificities, enzymes with altered activity, and improved biocatalysts to designing entirely new proteins. The former three examples are simpler in that they modify existing proteins by figuring out which amino acids to change. Completely de novo protein design (building entirely new proteins from scratch) promises a near unlimited palette of chemical reactions, biological interactions, and receptor cascades at our disposal. In fact, some AI algorithms have already demonstrated some success in this arena.
Twist Gene Fragments und Variant Libraries gehen solche Protein Engineering-Herausforderungen an
Protein Engineering-Anwendungen profitieren derzeit von der Twist Bioscience Produktsuite für die Proteinexpression und Mutagenese und werden verkörpert von den Bemühungen des Baker-Labors, neue Proteintherapeutika gegen Botulismus und Influenza zu entwickeln. Durch die Kombination von computergestützten Tools und Twist Oligo-Pools konnte das Baker-Team den Design-Build-Test-Zyklus für das De-novo-Protein Engineering erheblich beschleunigen. Twist Bioscience bietet auch Genfragmente, die den Klonierungsprozess für Proteinexpressionsexperimente in kleinem Maßstab vereinfachen, und Variant Libraries, die Hochdurchsatz-Analysen mutanter Proteine mit vollständiger Variantendarstellung ermöglichen.
Ob AlphaFold 2 die bestehenden Bemühungen im Bereich des Proteindesigns ergänzen oder das Inverse Proteinfaltungsproblem insgesamt lösen könnte, bleibt offen. AlphaFold 2 stimmte bei etwa einem Drittel der von CASP14 zugewiesenen Strukturen nicht mit der Leistung laborbasierter Methoden überein, was auf Verbesserungspotenzial hinweist. Trotzdem übertraf die Gesamtleistung von AlphaFold 2 die Konkurrenz und die des Vorgängers von 2018 bei weitem. Wenn dies die Leistungsverbesserung ist, die wir in nur zwei Jahren erwarten sollten, können wir uns nur vorstellen, wozu die nächste Iteration in der Lage sein wird.
Was denken Sie?
Gefällt mir
Gefällt mir nicht
Gefällt mir sehr
Überraschend
Interessant


{kind=link}