Les prédictions des protéines : de nouveaux outils pour comprendre ces structures complexes de la vie
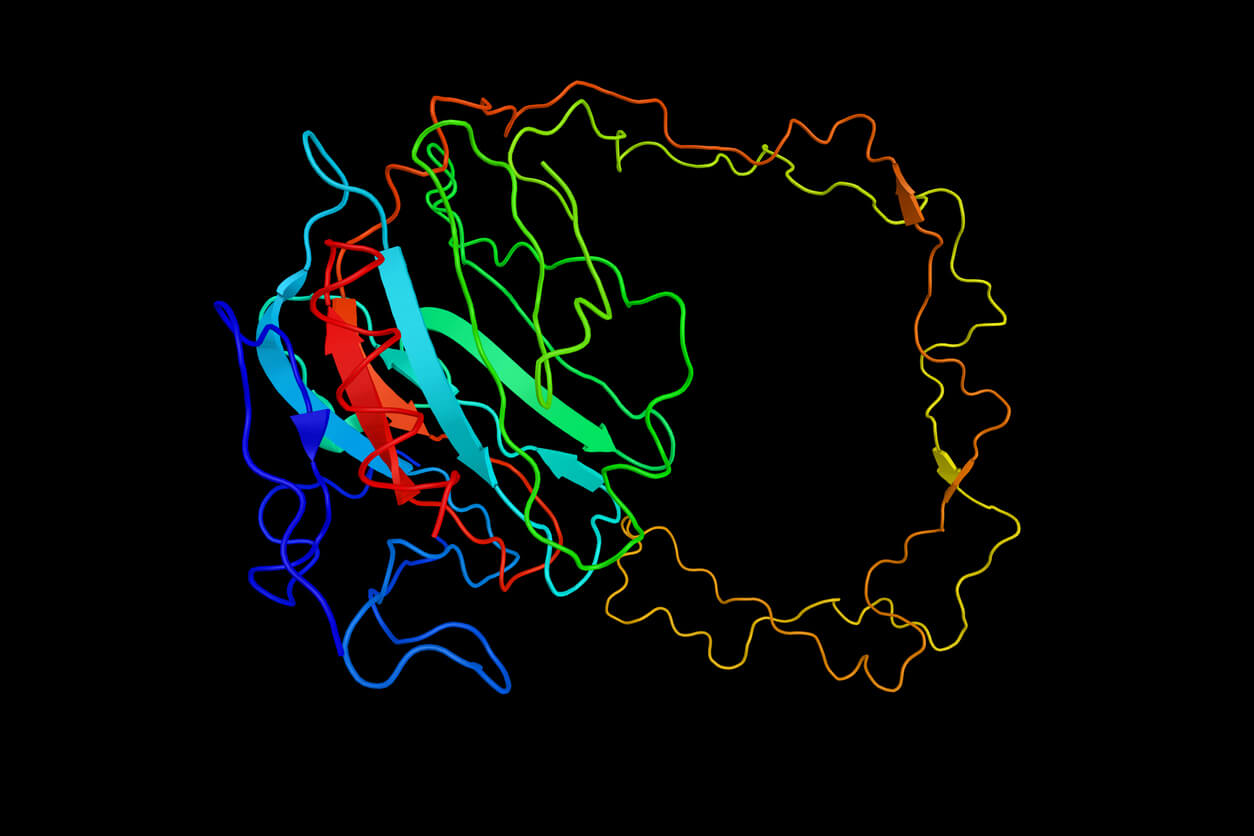
Les protéines sont les mécanismes moléculaires de la vie. Initialement constituées de chaînes d’acides aminés traduites à partir d’une séquence d’ARN correspondante, elles acquièrent leur fonctionnalité en se repliant en structures 3D complexes. La nature donne l’impression que le procédé est facile ; même avec des millions de configurations 3D possibles, la plupart des petites protéines se replient instantanément en une structure fonctionnelle stable grâce à diverses forces atomiques. Comprendre comment les protéines se replient est essentiel pour apporter les réponses recherchées en biologie. Par exemple, comment les mutations du génome entraînent-elles des maladies, ou comment une enzyme peut-elle être modifiée pour dégrader efficacement le plastique ?
La question ultime, un postulat vieux de 50 ans appelé « problème du repliement des protéines », consiste à savoir si la structure d’une protéine peut être prédite à partir de sa seule séquence d’acides aminés. Jusqu’à présent, la réponse a été un NON catégorique. Pour une chaîne d’acides aminés, il existe trop de structures possibles pour savoir laquelle est correcte.
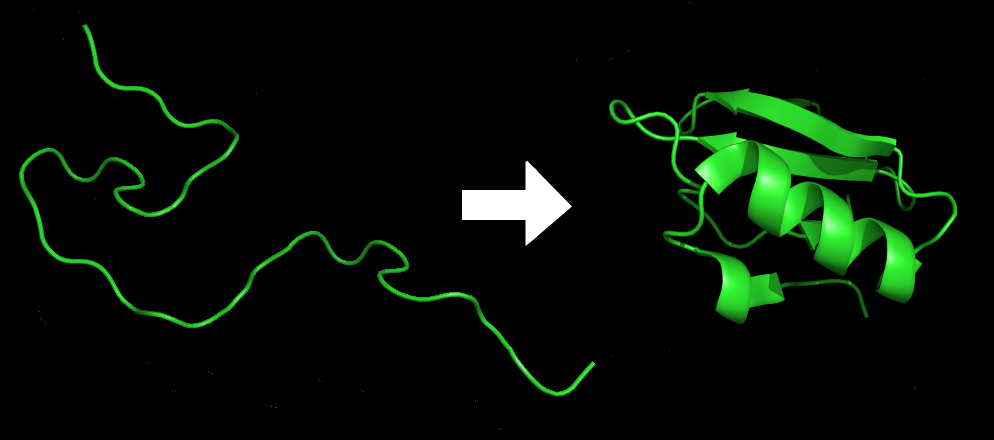
An amino acid chain for Chymotrypsin inhibitor 2 (1LW6) in its unfolded and folded form. Source: Wikicommons.
En général, la structure des protéines est déterminée par observation du diagramme de diffraction des rayons X qui traversent leurs formes cristallisées. Il est facile de faire éclater des cristaux de protéines avec des rayons X ; c’est la fabrication de ces cristaux qui est difficile. Nombreux sont ceux qui parlent d’un « art obscur », car il est compliqué de déterminer les conditions correctes pour provoquer la nucléation d’un cristal de protéine, et on y parvient généralement par la force pure ou par chance. Dans la littérature, on trouve même des exemples de cils errants contaminant une expérience, mais constituant le grain parfait pour la nucléation d’un cristal. Des méthodes alternatives comme la microscopie cryo-électronique et la spectroscopie par résonance magnétique nucléaire ont également été développées pour résoudre les structures des protéines. Cependant, toutes ces techniques de laboratoire sont à la fois coûteuses et longues.
Pour cette raison, les scientifiques ont essayé de mettre au point des méthodes informatiques permettant de résoudre les structures des protéines en se basant uniquement sur leur séquence d’acides aminés. Et malgré des efforts considérables, le domaine a été ponctué de petites avancées marginales dans ces méthodes pendant près de 50 ans, jusqu’à aujourd’hui.
Le concours du CASP annonce un gagnant
Tous les deux ans, une conférence est organisée pour évaluer les derniers développements en matière de résolution computationnelle de la structure des protéines. Appelée « Critical Assessment of Techniques for Protein Structure Prediction » (CASP), cette conférence bisannuelle met au défi des équipes de résoudre par le calcul des structures de protéines qui n’ont été déterminées que récemment par des méthodes de laboratoire.
At this year’s iteration, CASP14, Google’s DeepMind blew the competition away with its artificial intelligence platform AlphaFold 2. Not only did it handily outperform its competitors, it did as well as lab-based techniques for over two-thirds of the protein structures it was given to solve.
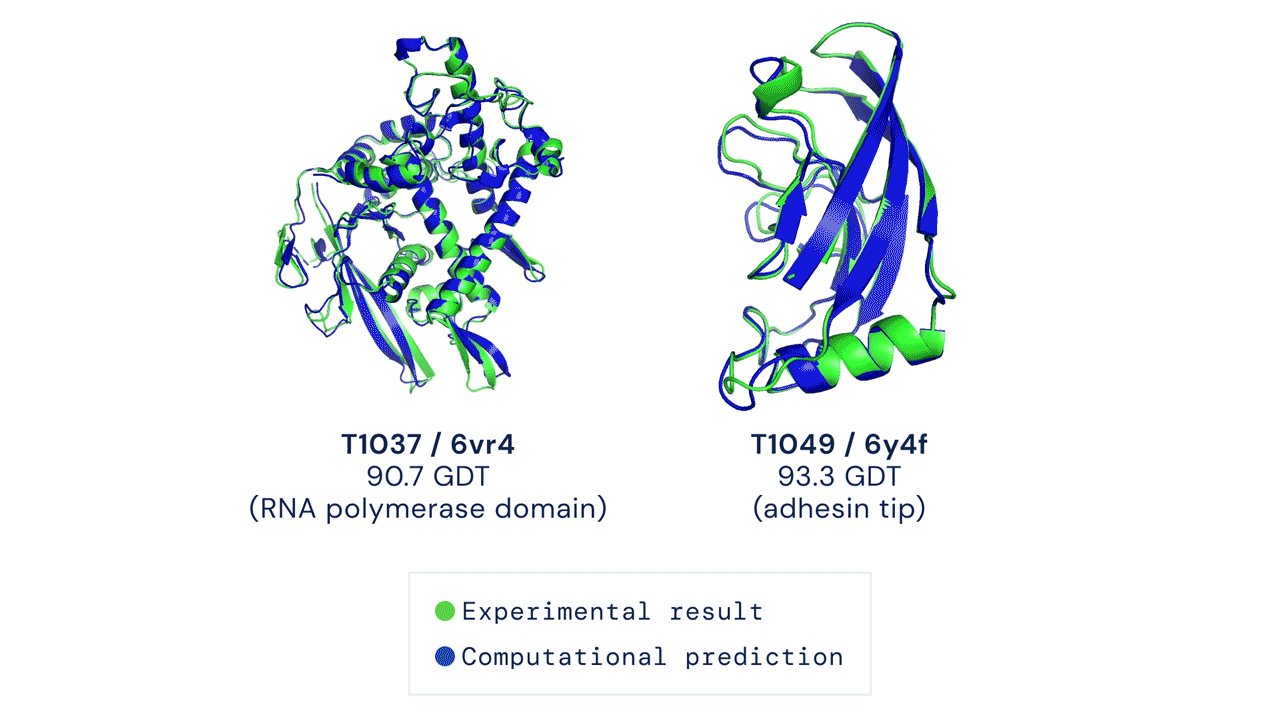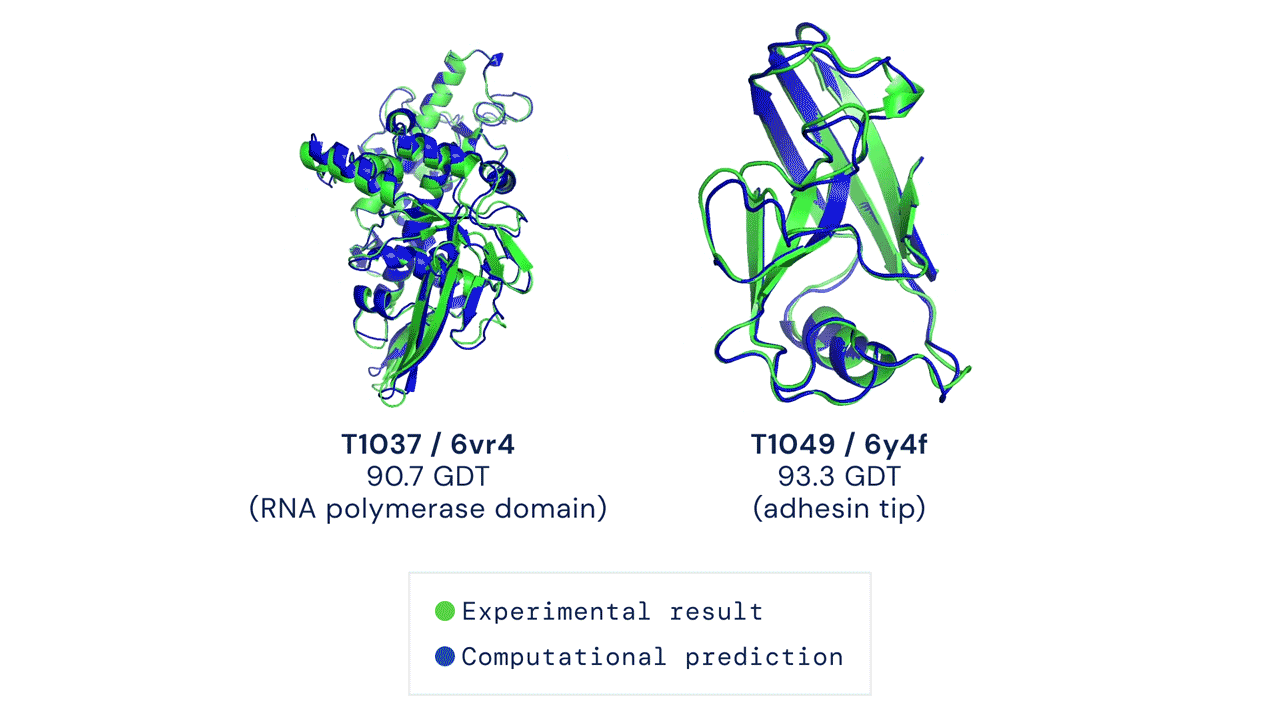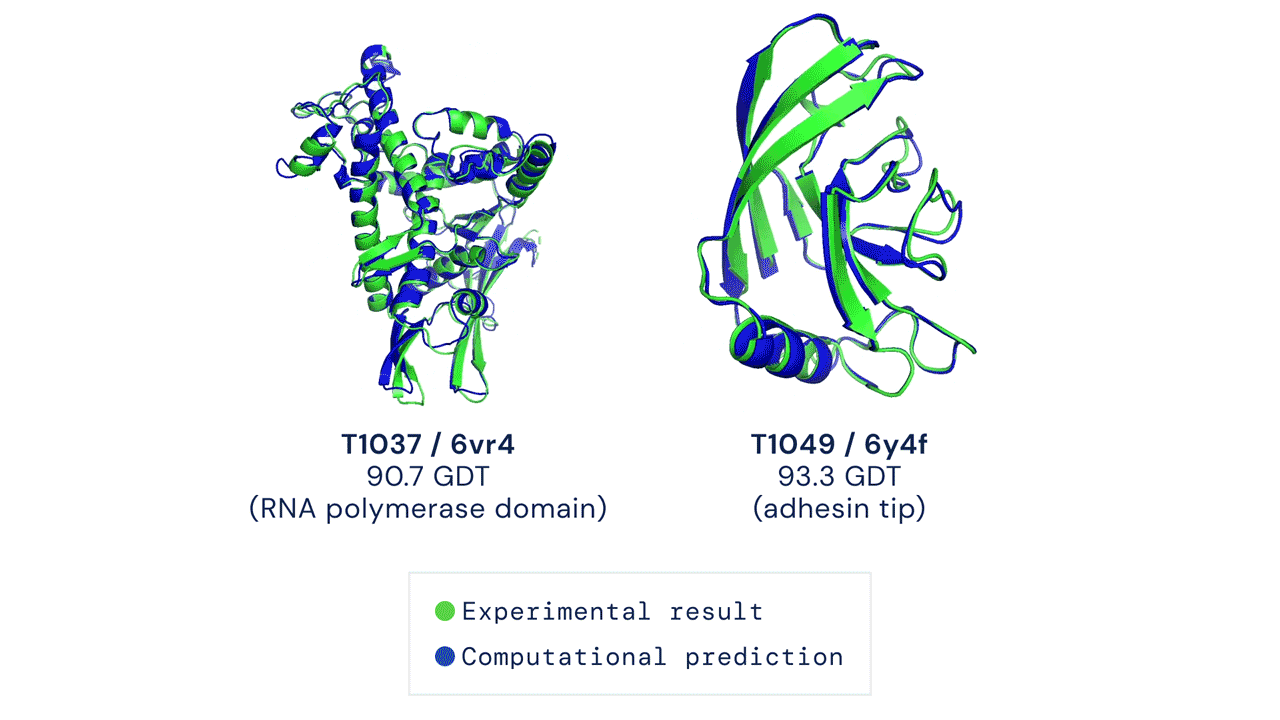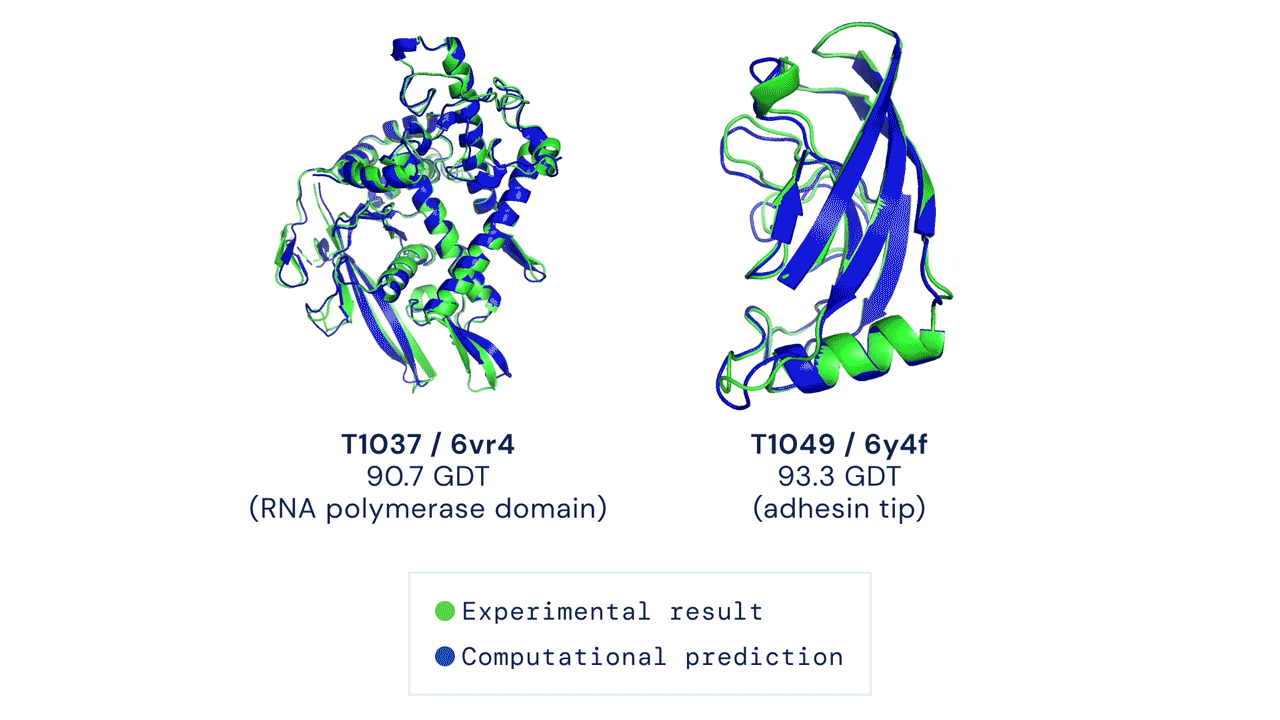
Deux structures protéiques prédites par AlphaFold ont détrôné leurs structures déduites expérimentalement. Source
Le CASP mesure la précision des algorithmes de repliement des protéines à l’aide d’une mesure appelée le test de distance globale (Global Distance Test, GDT). Sur une échelle de 0 à 100, le GDT évalue essentiellement la proximité entre une prédiction informatique et la structure déterminée en laboratoire. AlphaFold 2 a obtenu un score médian de 92,4 GDT dans CASP14. Un score aussi élevé n’aide pas à déterminer si la structure computationnelle ou empirique est plus « correcte ».
Under the Hood of AlphaFold 2
The AlphaFold algorithm uses deep learning to solve protein structures. Deep learning is a form of machine learning that performs a task by automatically extracting the features of a system (e.g. 3D structure) from raw input data (e.g. many protein structures and amino acid chains). Although the inner workings of AlphaFold 2 won’t be published until early 2021, what’s clear is that it uses a deep-learning technique called an attention network.
Une approche similaire sert à entraîner des machines à comprendre le langage. Pensez à cet article de blog comme à une longue chaîne d’acides aminés. Tout comme cette dernière forme des structures de protéines complexes par une série d’interactions locales et distantes entre les acides aminés, la première transmet du sens par des relations locales et distantes entre les mots, les phrases et les paragraphes. Les algorithmes d’apprentissage en profondeur basés sur l’attention synthétisent ces relations locales et distantes pour apprendre des tâches telles que la compréhension du sens du langage ou la détermination de la structure 3D d’une protéine.
En des termes moins abstraits, AlphaFold 2 a regroupé les informations provenant des séquences liées à l’évolution, des alignements de séquences multiples et des interactions entre les paires de résidus d’acides aminés pour apprendre comment ces variables sont liées à travers les 170 000 structures de protéines connues utilisées pour entraîner l’algorithme.
AlphaFold 2 peut-il résoudre des problèmes du monde réel ?
Sa capacité à résoudre des structures en quelques semaines plutôt qu’en plusieurs mois (ou années) laisse entrevoir un rôle de cette plateforme d’IA dans la découverte de petites molécules thérapeutiques et l’ingénierie de nouvelles protéines pour une variété d’applications du monde réel, de l’optimisation des processus industriels à la dégradation du plastique.
Computational methods are frequently used to determine how drugs interact with their protein targets. Unfortunately, the structure of thousands of druggable proteins remains unknown, making it difficult to develop effective drugs against them. Predictive algorithms like AlphaFold 2 are promising solutions to this backlog if shown to be consistently accurate. In a proof-of-principle demonstration, AlphaFold 2 accurately predicted the structure of the SARS-CoV-2 Spike protein, the main therapeutic target against COVID-19, earlier this year. It also predicted several other SARS-CoV-2 proteins before they were confirmed by lab-based methods.
Another game-changing application where AlphaFold 2 may make its mark is in protein engineering. Often called the inverse protein-folding problem, protein design aims to identify amino acid sequences that will form stabilized, functional, useful protein structures. It encapsulates everything from engineering receptors with different ligand specificities, enzymes with altered activity, and improved biocatalysts to designing entirely new proteins. The former three examples are simpler in that they modify existing proteins by figuring out which amino acids to change. Completely de novo protein design (building entirely new proteins from scratch) promises a near unlimited palette of chemical reactions, biological interactions, and receptor cascades at our disposal. In fact, some AI algorithms have already demonstrated some success in this arena.
Les fragments de gènes de Twist Bioscience et les banques de variants facilitent ces défis d’ingénierie des protéines
Les applications d’ingéniérie des protéines bénéficient actuellement de la gamme de produits Twist Bioscience pour l’expression des protéines et la mutagenèse. Ces activités sont illustrées par les travaux du laboratoire Baker visant à concevoir de nouvelles protéines thérapeutiques contre le botulisme et la grippe. En combinant les outils informatiques et les pools d’oligonucléotides de Twist Bioscience, l’équipe de Baker a pu considérablement accélérer le cycle conception-construction-test de l’ingénierie des protéines de novo. Twist Bioscience propose également des fragments de gènes qui simplifient le processus de clonage pour les expériences d’expression de protéines à petite échelle et des Banques de variants qui permettent un criblage à haut débit de protéines mutantes avec une représentation complète des variants.
La question de savoir si AlphaFold 2 pourrait contribuer aux efforts existants en matière de conception de protéines, ou résoudre le problème du repliement inverse des protéines, reste ouverte. AlphaFold 2 n’a pas atteint la performance des méthodes de laboratoire pour environ un tiers des structures définies par CASP14, ce qui indique que des progrès restent à faire. Néanmoins, les performances globales d’AlphaFold 2 ont largement dépassé celles de ses concurrents et celles de son prédécesseur de 2018. Si telle est l’amélioration des performances à laquelle nous devons nous attendre en seulement deux ans, alors nous ne pouvons qu’imaginer ce dont la prochaine itération sera capable.
Qu’en pensez-vous ?
J’aime
bien
6
Je n’aime pas
0
J’aime beaucoup
1
Je suis surpris(e)
0
C’est intéressant
1


{kind=link}