









RNA sequencing (RNA-seq) has emerged as a powerful tool in the molecular biologist’s toolkit. Whereas DNA sequencing can be used to make predictions about cell biology, RNA-seq enables researchers to follow information through life’s central dogma (DNA to RNA to protein), detailing the dynamic nature of gene expression and revealing the nuanced patterns that drive cell behavior and disease.
However, as with DNA sequencing, realizing the full potential of RNA-seq can be a tricky task. RNA degradation is a problem under the best of conditions, but is a particular issue when working with clinical samples, which are often subject to the destructive effects of formalin-fixation and paraffin-embedding. Collecting informative RNA-seq data from these samples is hard enough, but doing so with sensitivity and efficiency can frustrate even the best of us.
Because of these challenges, designing and optimizing RNA-seq experiments can be a significant bottleneck for most researchers, particularly when working in isolation. But, with quality library preparation, well-designed target capture, and simple workflows, RNA-seq can become just another unremarkable process that produces remarkable data.
A comprehensive, accessible solution to RNA-seq
Twist has made it easy for researchers to get expert help through a suite of comprehensive RNA-seq solutions, ranging from optimized library preparation kits to expert-designed and customized target enrichment panels. When combined, these tools enable a streamlined RNA-seq workflow that can be used for a range of applications, from differential gene expression analyses to gene-fusion detection and biomarker discovery.
Efficiency in Whole Transcriptome RNA-seq Through Depletion
There are many different approaches to RNA-seq, and which one you choose will depend on the application. For example, if your aim is to carry out deep profiling of various different RNA species (such as mRNA, lncRNA, and precursor RNA), you’re likely to employ whole transcriptome sequencing—an approach that casts a broad net, capturing and sequencing RNA molecules without bias.
Whole transcriptome sequencing can be favored when discovery or data banking is the goal. By not focusing on any single transcript type, whole transcriptome sequencing allows for the discovery of unexpected transcripts, but this comes at the cost of generating an overwhelming amount of data, including transcripts that may be irrelevant to specific research goals.
To reduce the cost of whole transcriptome sequencing, it's often necessary to deplete the total RNA pool of highly expressed ribosomal RNA (rRNA) and, when necessary, hemoglobin transcripts during library preparation. Without depletion, the high-abundance transcripts may consume a substantial amount of sequencing resources, reducing the depth of sequencing over informative transcripts and diminishing the overall sensitivity of the assay. Researchers thus have the challenge of removing high-abundance transcripts without accidentally losing desired RNA.
To solve this problem, Twist offers a library preparation workflow that enables highly specific depletion of rRNA and hemoglobin transcripts. Relative to similar library preparation workflows, Twist’s solution is approximately 6 fold more effective. This means researchers achieve substantially better depletion, empowering them to perform more sensitive and efficient whole transcriptome sequencing.
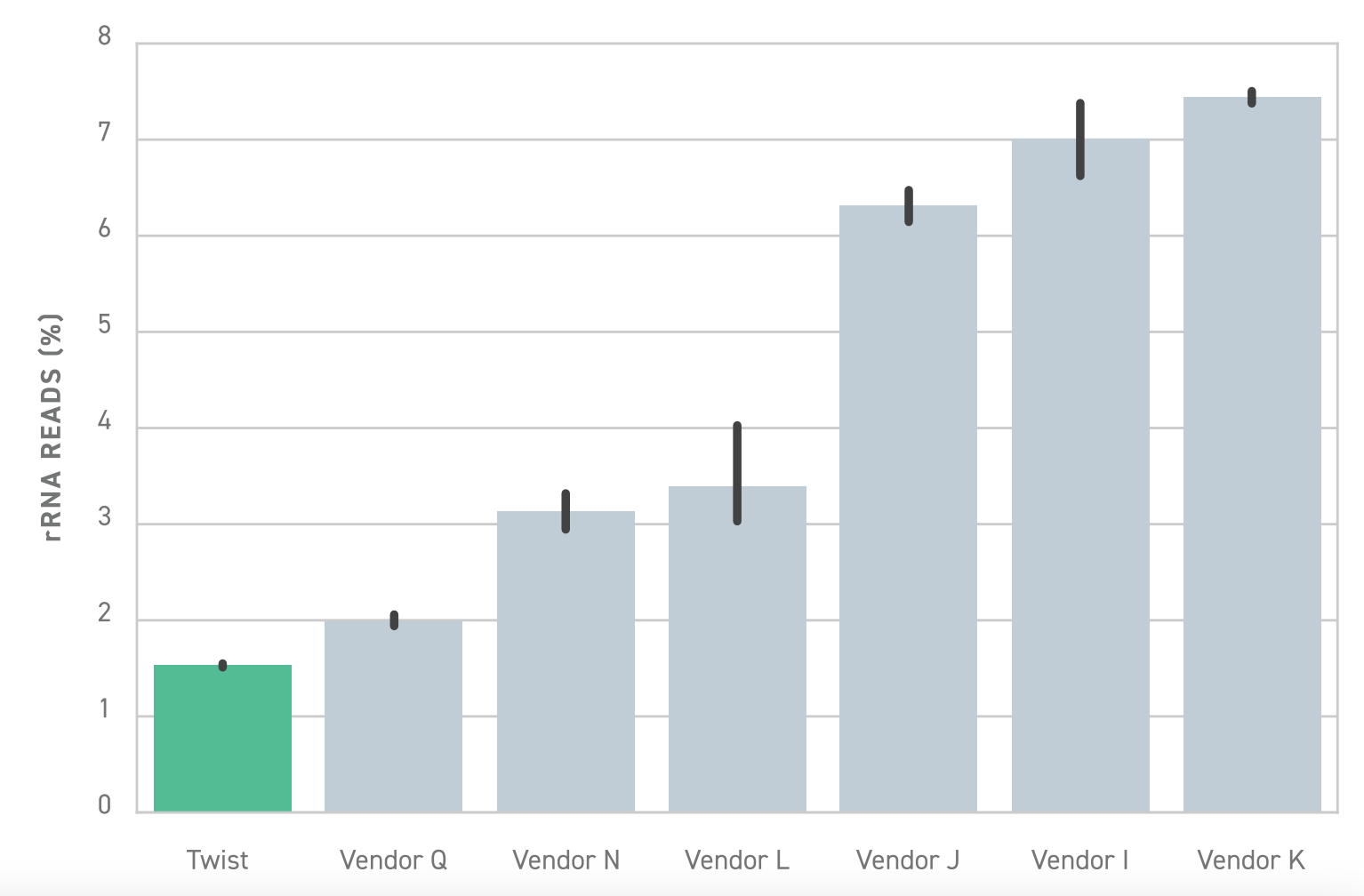
In some cases where only a select few transcripts are of interest, targeted RNA sequencing may be a more cost-effective choice over whole transcriptome sequencing.
Achieving precise and uniform target-enriched RNA-seq
Target enrichment is a common approach to RNA-seq. Rather than spending precious time and resources sequencing a broad collection of RNA transcripts, researchers can selectively enrich their sample libraries to only contain the transcripts researchers are interested in. By doing this, sequencing resources can be focused on just those transcripts of interest, enabling deeper sequencing and, consequently, more sensitive transcript detection.
Enrichment can be done through one of two methods (amplicon or hybrid-capture). Both approaches have their merits, but in recent years hybrid-capture methods have led the way. Here, oligonucleotide probes that are complementary to the target transcripts are synthesized with a biotin conjugate attached. When incubated with the sample library, these probes bind to their target transcripts and can be subsequently isolated using streptavidin-coated magnetic beads. Such an approach allows researchers to precisely isolate target transcripts and greatly improve the efficiency and sensitivity of their assay.
The success of target capture is heavily influenced by the quality of the capture probe library that’s used, both in its design and synthesis. To this end, Twist provides a comprehensive collection of tools to support researchers in targeted RNA-seq. Custom target capture panels can be designed with guidance from Twist’s team of design experts to maximize capture efficiency.
While valuable for many applications—such as gene signature analysis, gene fusion detection, and focused differential gene expression studies—targeted RNA-seq is often limited in its discovery potential. In designing capture probes, the targeted approach is inherently biased for transcripts that you expect to find (and capture). Therefore targeted RNA-seq has historically not been well suited for biomarker discovery or the detection of unexpected RNA fusion and spliceform transcripts.

Improving targeted RNA-seq for discovery
To help improve the discovery power of targeted RNA-seq, Twist has developed a specialized RNA target enrichment panel with a design that enables the hypothesis-free capture of protein-coding transcripts. This panel, known as the Twist RNA Exome, uses a capture probe design that facilitates the capture of unexpected transcripts, including alternatively spliced transcripts and fusion genes.

Typically, targeted RNA-seq approaches struggle to detect aberrant or rare spliceoform transcripts because capture probes are tiled across exon-exon junctions. This creates a bias for transcripts in which the expected exon-exon junctions are intact. In contrast, The Twist RNA Exome utilizes an exon-aware algorithm to ensure that capture probes remain within exon boundaries. As such, wherever targeted exons may be, they can be captured, even if they are located in an abnormal transcript.
The benefits of Twist’s exon-aware approach extend beyond aberrant transcripts. Results suggest that overall enrichment with the exon-aware design strategy is significantly improved over a conventional tiling design and delivers a significant increase in signal over whole transcriptome sequencing with the same number of reads. This leads to a substantial increase in the number of detected coding genes, even when using RNA from formalin-fixed and paraffin-embedded samples. Importantly, this improvement enables the detection of low-expressing and rare transcripts.
In effect, the Twist RNA Exome combines the benefits of sequencing approaches into one package, preserving the sensitivity and efficiency gained from target enrichment and the discovery power of whole transcriptome sequencing.
A Simplified, Efficient, and Sensitive Workflow

Twist’s RNA sequencing portfolio is built to empower researchers, whatever your goals may be, with efficient and sensitive RNA-seq workflows. Importantly, these tools have all been designed in tandem with one another, meaning that everything from the Twist Library Prep kit to the Twist RNA Exome have been optimized for use with one another—ensuring that researchers are set up for success. RNA-seq is never simple, but with Twist, you can make it look easy.
🧬 Our Superior Library Preparation
Library preparation is a critical step in any RNA-sequencing project. The basic premise is to isolate the desired RNA molecules from your sample, convert them to a format that’s compatible with sequencing, and attach identifiers that enable higher-resolution downstream analyses. The specifics of library preparation can differ depending on your application, so it's important to find a kit that fits your needs.
To enable sensitive and robust RNA sequencing across a wide range of applications, Twist has developed a comprehensive suite of library preparation solutions that save time and sequencing costs while optimizing for performance in any workflow. Specifically, these solutions are designed with the following features to enable high-quality RNA-seq:
- Ease of use and speed: Whereas most library preparation kits require 6-8 hours, Twist’s RNA-seq library preparation can be done in less than 5 hours.
- Sensitivity for a wide range of inputs: Twist RNA Library preparation accepts input from 1 ng to 1 000 ng of RNA, enabling data collection from myriad sample types.
- Compatibility with low-quality RNA: With the ability to incorporate a repair step, Twist’s RNA library preparation enables the sequencing of RNA from difficult sample types, including FFPE.
- Comprehensive: Twist offers RNA library preparation for both whole transcriptome and targeted RNA sequencing by incorporating depletion or positive selection steps, depending on your needs.

*Réservé à l’utilisation dans le cadre de la recherche. Not for use in diagnostic procedures.
Qu’en pensez-vous ?
J’aime
bien
Je n’aime pas
J’aime beaucoup
Je suis surpris(e)
C’est intéressant

