Predicciones de proteínas: Nuevas herramientas para comprender estas estructuras complejas de la vida
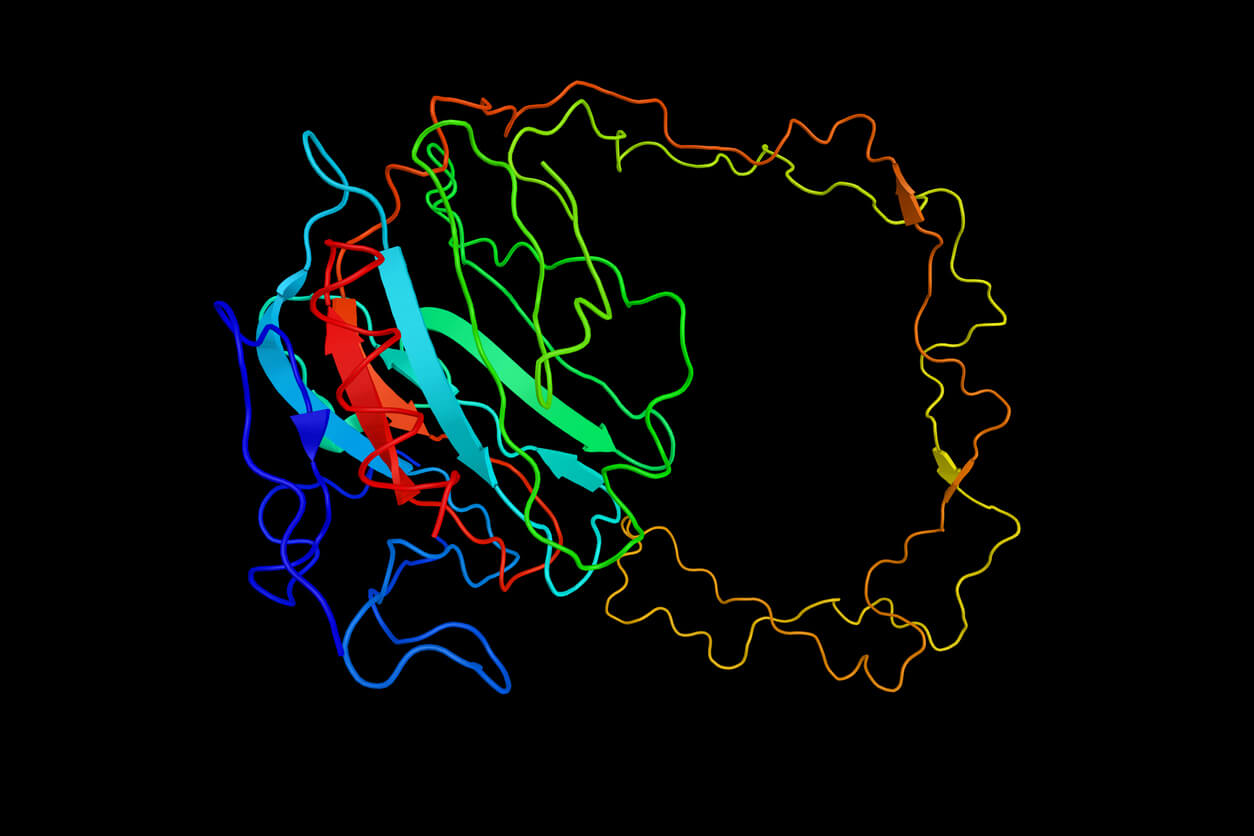
Las proteínas son las máquinas moleculares de la vida. Empiezan como cadenas de aminoácidos traducidas a partir de una secuencia de ARN correspondiente y adquieren su funcionalidad plegándose en estructuras complejas 3D. La naturaleza hace que este proceso parezca fácil; incluso con millones de configuraciones 3D posibles, la mayoría de las proteínas pequeñas se pliegan en una estructura funcional estable de forma instantánea a través de diversas fuerzas atómicas. La comprensión de cómo funciona el plegamiento de las proteínas es clave para revelar muchas de las respuestas buscadas en biología. Por ejemplo, ¿cómo las mutaciones del genoma provocan la enfermedad o cómo puede diseñarse una enzima para degradar plástico de forma efectiva?
La pregunta definitiva, un postulado que ahora tiene 50 años denominado “el problema del plegamiento de la proteína”, plantea si se puede predecir la estructura proteica utilizando únicamente su secuencia de aminoácidos. Hasta ahora, la respuesta ha sido un NO contundente. Con una cadena de aminoácidos dada, hay demasiadas estructuras posibles como para saber cuál es la correcta.
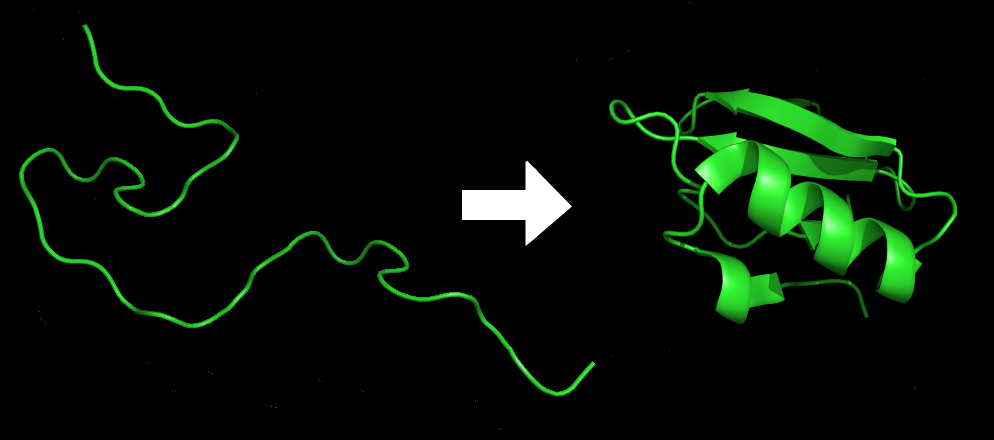
An amino acid chain for Chymotrypsin inhibitor 2 (1LW6) in its unfolded and folded form. Source: Wikicommons.
Normalmente, las estructuras proteicas se determinan mediante la observación del patrón de difracción de los rayos X al pasar por sus formas cristalizadas. Atravesar los cristales de proteínas con rayos X es sencillo; lo que es difícil es fabricar dichos cristales. Muchos lo denominan “magia negra”, ya que identificar las condiciones correctas para provocar la nucleación de un cristal de proteínas es, una vez más, complejo, y normalmente se consigue mediante la fuerza bruta o pura suerte. En la bibliografía, incluso pueden encontrarse ejemplos de pestañas caídas que contaminaron un experimento, pero fueron la semilla perfecta para la nucleación de los cristales. También se han desarrollado otros métodos, como la microscopía electrónica criogénica y la espectroscopía de resonancia magnética nuclear, para resolver estructuras proteicas. Sin embargo, todas estas técnicas basadas en el laboratorio son caras y laboriosas.
Este es el motivo por el que los científicos han estado intentando concebir métodos computacionales para resolver estructuras proteicas basándose únicamente en su secuencia de aminoácidos. Y, a pesar de sus grandes esfuerzos, solo se han conseguido pequeños avances incrementales en estos métodos durante casi 50 años… hasta ahora.
La competición CASP anuncia un ganador
Cada dos años, se celebra una conferencia para evaluar los últimos desarrollos en la resolución computacional de estructuras proteicas. Denominada evaluación crítica de las técnicas para la predicción de estructuras proteicas (CASP), la conferencia bianual reta a los equipos a resolver computacionalmente estructuras proteicas que recientemente se han determinado con métodos basados en laboratorio.
At this year’s iteration, CASP14, Google’s DeepMind blew the competition away with its artificial intelligence platform AlphaFold 2. Not only did it handily outperform its competitors, it did as well as lab-based techniques for over two-thirds of the protein structures it was given to solve.
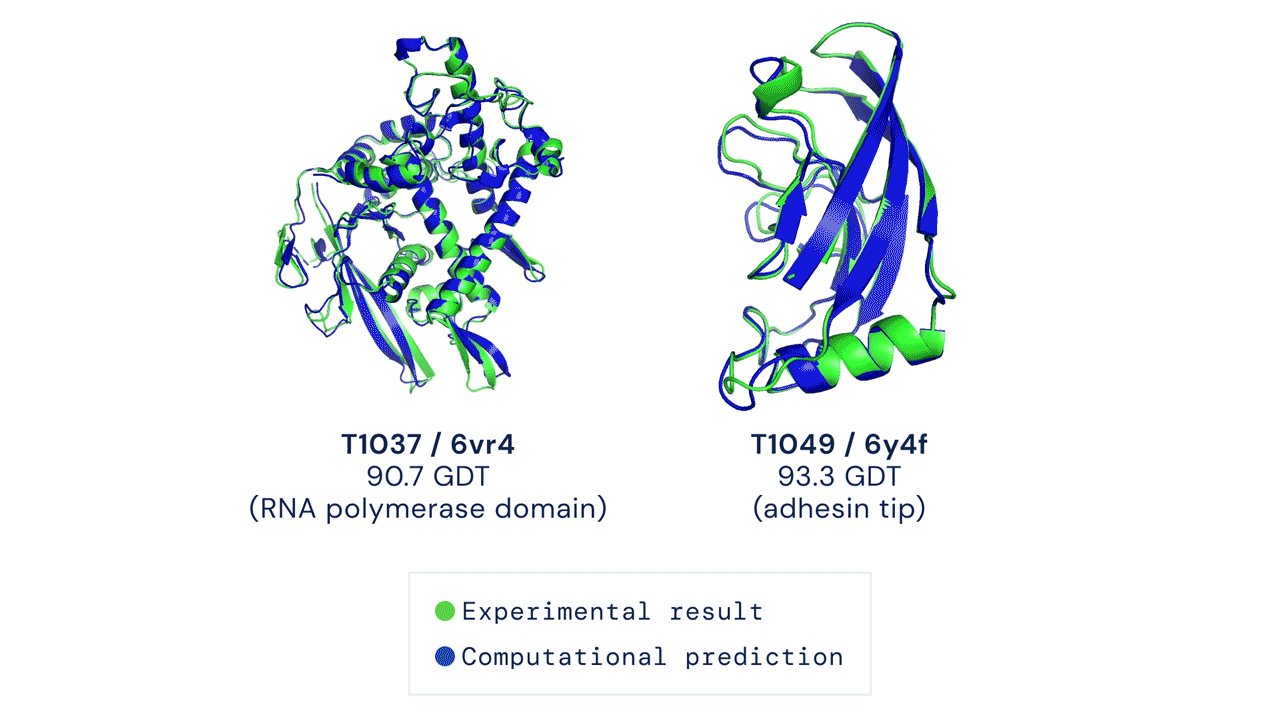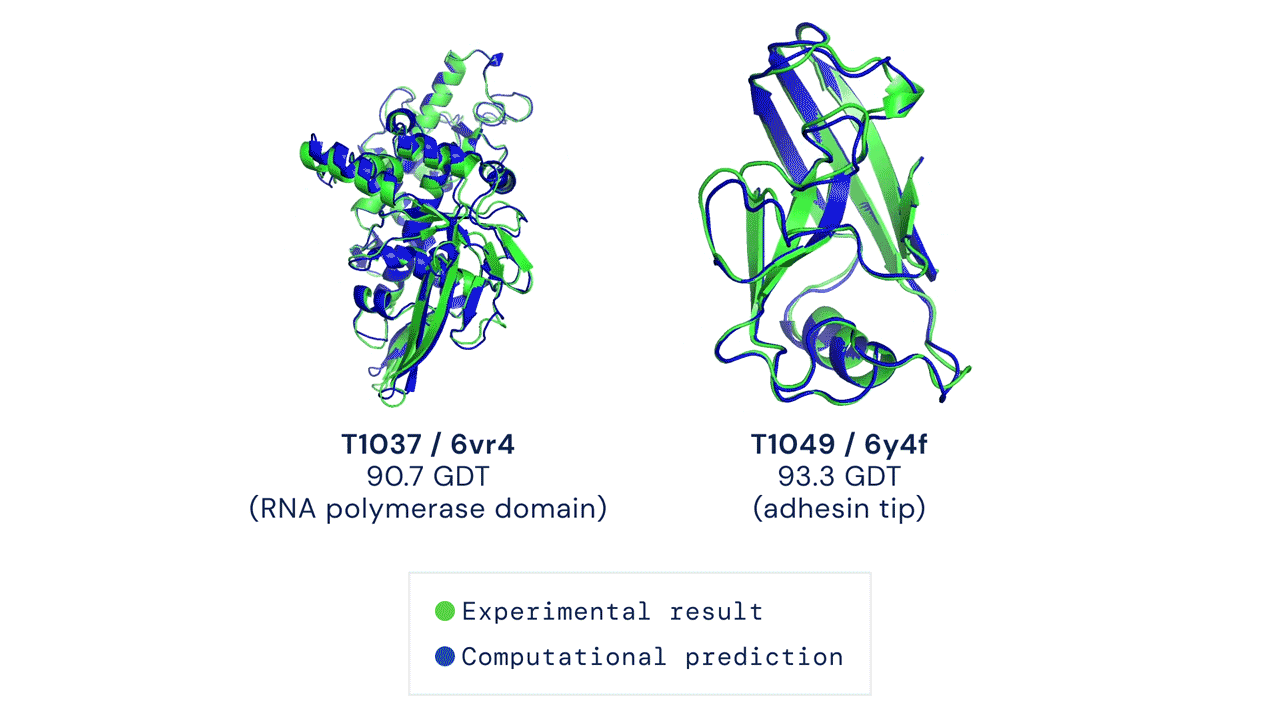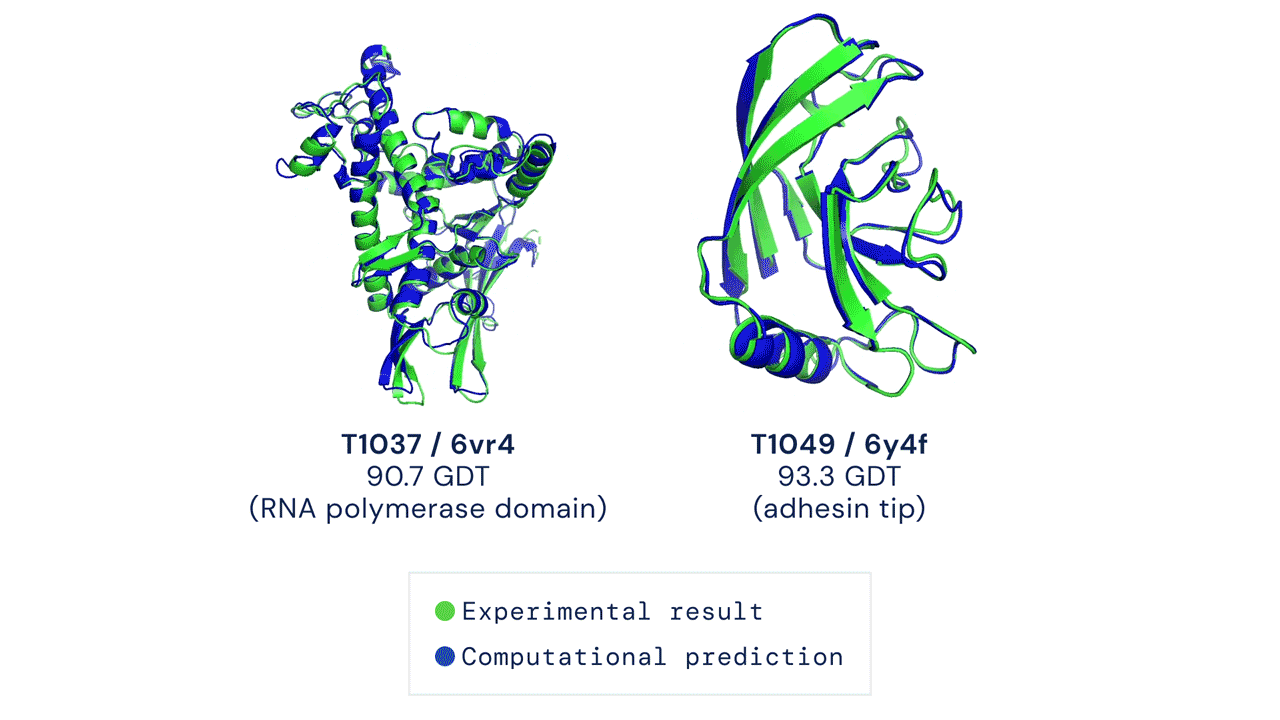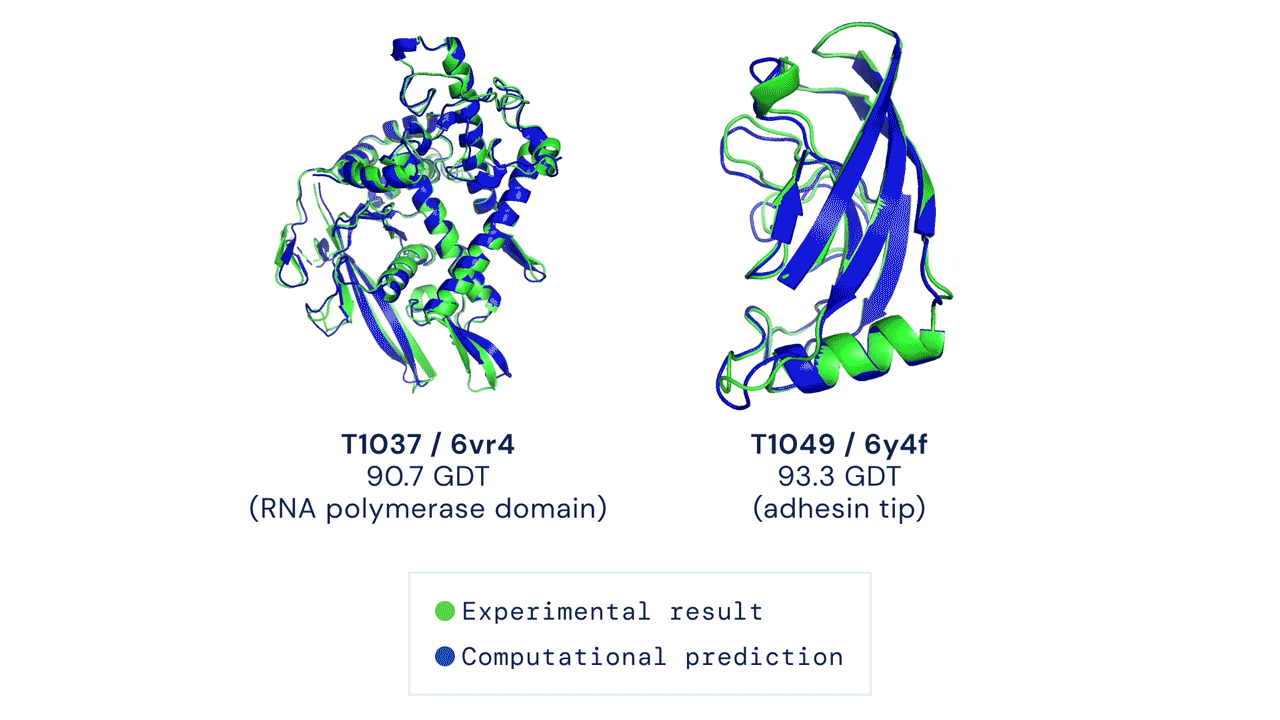
Dos estructuras proteicas predichas por AlphaFold superpuestas sobre sus estructuras deducidas experimentalmente. Fuente
La CASP mide la precisión de los algoritmos de plegamiento de proteínas con una métrica denominada la prueba de distancia global (GDT). Con una puntuación de 0 a 100, la GDT evalúa en esencia cómo de cerca está una predicción computacional de la estructura determinada en el laboratorio. AlphaFold 2 obtuvo una mediana de 92,4 GDT en la CASP14. Una puntuación tan alta hace que sea difícil precisar cuál es la estructura más “correcta”, la computacional o la empírica.
Under the Hood of AlphaFold 2
The AlphaFold algorithm uses deep learning to solve protein structures. Deep learning is a form of machine learning that performs a task by automatically extracting the features of a system (e.g. 3D structure) from raw input data (e.g. many protein structures and amino acid chains). Although the inner workings of AlphaFold 2 won’t be published until early 2021, what’s clear is that it uses a deep-learning technique called an attention network.
Se utiliza un enfoque similar para enseñar a las máquinas a comprender el idioma. Considere que este artículo de blog es una larga cadena de aminoácidos. Al igual que esta última forma estructuras proteicas complejas a través de una serie de interacciones locales y distantes entre los aminoácidos, el artículo expresa su significado mediante relaciones locales y distantes entre palabras, sentencias y párrafos. Los algoritmos de aprendizaje profundo basados en la atención sintetizan estas relaciones locales y distantes para aprender tareas, como la comprensión del significado del lenguaje o la determinación de la estructura 3D de una proteína.
En términos menos abstractos, AlphaFold 2 combinó la información derivada de secuencias relacionadas evolutivamente, varias alineaciones de secuencias e interacciones entre pares de residuos de aminoácidos para descubrir cómo estas variables se relacionan en las 170 000 estructuras proteicas conocidas utilizadas para entrenar al algoritmo.
¿Puede AlphaFold 2 solucionar problemas del mundo real?
Su capacidad para resolver estructuras en semanas en lugar de en meses (o años) indica un papel para la plataforma de IA en el descubrimiento de moléculas pequeñas terapéuticas y la ingeniería de nuevas proteínas para diversas aplicaciones del mundo real, desde la optimización de procesos industriales hasta la degradación de plásticos.
Computational methods are frequently used to determine how drugs interact with their protein targets. Unfortunately, the structure of thousands of druggable proteins remains unknown, making it difficult to develop effective drugs against them. Predictive algorithms like AlphaFold 2 are promising solutions to this backlog if shown to be consistently accurate. In a proof-of-principle demonstration, AlphaFold 2 accurately predicted the structure of the SARS-CoV-2 Spike protein, the main therapeutic target against COVID-19, earlier this year. It also predicted several other SARS-CoV-2 proteins before they were confirmed by lab-based methods.
Another game-changing application where AlphaFold 2 may make its mark is in protein engineering. Often called the inverse protein-folding problem, protein design aims to identify amino acid sequences that will form stabilized, functional, useful protein structures. It encapsulates everything from engineering receptors with different ligand specificities, enzymes with altered activity, and improved biocatalysts to designing entirely new proteins. The former three examples are simpler in that they modify existing proteins by figuring out which amino acids to change. Completely de novo protein design (building entirely new proteins from scratch) promises a near unlimited palette of chemical reactions, biological interactions, and receptor cascades at our disposal. In fact, some AI algorithms have already demonstrated some success in this arena.
Los fragmentos de gen Twist y las bibliotecas de variantes facilitan dichos retos de la ingeniería de proteínas
Actualmente, las aplicaciones de ingeniería de proteínas se benefician del conjunto de productos de Twist para la expresión de proteínas y la mutagénesis. Estos esfuerzos están representados por los esfuerzos del laboratorio Baker para diseñar nuevos tratamientos con proteínas frente al botulismo y la gripe. Al combinar las herramientas computaciones y los grupos de oligonucleótidos Twist, el equipo de Baker pudo acelerar enormemente el ciclo de diseño-creación-comprobación de la ingeniería de proteínas de novo. Twist también ofrece fragmentos de gen que simplifican los procesos de clonación para experimentos de expresión de proteínas a pequeña escala y bibliotecas de variantes que permiten un cribado de alto rendimiento de proteínas mutantes con una representación completa de las variantes.
Si AlphaFold 2 podría complementar los esfuerzos existentes en el diseño de proteínas o solucionar completamente el problema del plegamiento inverso de la proteína siguen siendo preguntas abiertas. AlphaFold 2 no alcanzó el rendimiento de los métodos de laboratorio para aproximadamente un tercio de las estructuras asignadas por la CASP14, lo que indica margen de mejora. Sin embargo, el rendimiento general del AlphaFold 2 ha sobrepasado de lejos el de sus competidores y el de su antecesor de 2018. Si esta es la mejora en el rendimiento que deberíamos esperar en tan solo dos años, solo podemos imaginar lo que podrá hacer la próxima versión.
¿Qué piensa?
Me gusta
No me gusta
Me encanta
Me asombra
Me interesa


{kind=link}